16 Accessing Ancient Metagenomic Data
For this chapter’s exercises, if not already performed, you will need to download the chapter’s dataset, decompress the archive, and create and activate the conda environment.
Do this, use wget or right click and save to download this Zenodo archive: 10.5281/zenodo.17155430, and unpack
tar xvf accessing-ancient-metagenomic-data.tar.gz
cd accessing-ancient-metagenomic-data/You can then create the subsequently activate environment with
conda env create -f accessing-ancient-metagenomic-data.yml
conda activate accessing-ancient-metagenomic-data16.1 Introduction
In most bioinformatic projects, we need to include publicly available comparative data to expand or compare our newly generated data with.
Including public data can benefit ancient metagenomic studies in a variety of ways. It can help increase our sample sizes (a common problem when dealing with rare archaeological samples) - thus providing stronger statistical power. Comparison with a range of previously published data of different preservational levels can allow an estimate on the quality of the new samples. When considering solely (re)using public data, we can consider that this can also spawn new ideas, projects, and meta analyses to allow further deeper exploration of ancient metagenomic data (e.g., looking for correlations between various environmental factors and preservation).
Fortunately for us, geneticists and particularly palaeogenomicists have been very good at uploading raw sequencing data to well-established databases (Anagnostou et al. 2015).
In the vast majority of cases you will be able to find publically available sequencing data on the INSDC (https://www.insdc.org/) association of databases, namely the EBI’s European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/), and NCBI (https://www.ncbi.nlm.nih.gov/sra) or DDBJ’s (https://www.ddbj.nig.ac.jp/dra/index-e.html) Sequence Read Archives (SRA). However, you may in some cases find ancient metagenomic data on institutional FTP servers, domain specific databases (e.g. OAGR (https://oagr.org), Zenodo (https://zenodo.org), Figshare (https://figshare.com), or GitHub (https://github.com)).
But while the data is publicly available, we need to ask whether it is ‘FAIR’.
16.2 Finding Ancient Metagenomic Data
FAIR principles (Wilkinson et al. 2016) were defined by researchers, librarians, and industry in 2016 to improve the quality of data uploads - primarily by making data uploads more ‘machine readable’. FAIR standards for:
- Findable
- Accessible
- Interoperable
- Reusable
When we consider ancient (meta)genomic data, we are pretty close to this. Sequencing data is in most cases accessible (via the public databases like ENA, SRA), interoperable and reproducible because we use field standard formats such as FASTQ or BAM files. However findable remains an issue.
This is because the metadata about each data file is dispersed over many places, and very often not with the data files themselves.
In this case I am referring to metadata such as: What is the sample’s name? How old is it? Where is it from? Which enzymes were used for library construction? What sequencing machine was this library sequenced on?
To find this information about a given data file, you have to search many places (main text, supplementary information, the database itself), for different types of metadata (as authors report different things), and also in different formats (text, tables, figures).
This very heterogenous landscape makes it difficult for machines to index all this information (if at all), and thus means you cannot search for the data you want to use for your own research in online search engines.
16.3 AncientMetagenomeDir
This is where the SPAAM community project ‘AncientMetagenomeDir’ (https://github.com/spaam-community/AncientMetagenomeDir) comes in (Fellows Yates, Andrades Valtueña, et al. 2021). AncientMetagenomeDir is a resource of lists of metadata of all publishing and publicly available ancient metagenomes and microbial genome-level enriched samples and their associated libraries.
By aggregating and standardising metadata and accession codes of ancient metagenomic samples and libraries, the project aims to make it easier for people to find comparative data for their own projects, appropriately re-analyse libraries, as well as help track the field over time and facilitate meta analyses.
Currently the project is split over three main tables: host-associated metagenomes (e.g. ancient microbiomes), host-associated single-genomes (e.g. ancient pathogens), and environmental metagenomes (e.g. lakebed cores or cave sediment sequences).
The repository already contains more than 2000 samples and 5000 libraries, spanning the entire globe and as far back as hundreds of thousands of years.
To make the lists of samples and their metadata as accessible and interoperable as possible, we utilise simple text (TSV - tab separate value) files - files that can be opened by pretty much all spreadsheet tools (e.g., Microsoft Office excel, LibreOffice Calc) and languages (R, Python etc.) (Figure 16.1).

Critically, by standardising the recorded all metadata across all publications this makes it much easier for researchers to filter for particular time periods, geographical regions, or sample types of their interest - and then use the also recorded accession numbers to efficiently download the data.
At their core all different AncientMetagenomeDir tables must have at 6 minimum metadata sets at the sample level:
- Publication information (doi)
- Sample name(s)
- Geographic location (e.g. country, coordinates)
- Age
- Sample type (e.g. bone, sediment, etc.)
- Data Archive and accessions
Each table then has additional columns depending on the context (e.g. what time of microbiome is expected for host-associated metagenomes, or species name of the genome that was reconstructed).
The AncientMetagenomeDir project already has 12 major releases, and will continued to be regularly updated as the community continues to submit new metadata of samples of new publications as they come out.
What is the naming scheme of the AncientMetagenomeDir releases? Try to find the ‘release’ and ‘wiki’ sections of the GitHub repository interface and see if you can find the information…
The ‘release’ listing of a GitHub repository can be found on the right hand metadata bar.
In many cases you can also find a ‘CHANGELOG’ file that will list all the changes that have been made on each release.
The release name scheme is of different places listed on the Unesco World Heritage list (https://whc.unesco.org/en/list/). More background you can find here on the Wiki page (https://github.com/SPAAM-community/AncientMetagenomeDir/wiki/Release-Name-List)
16.4 AMDirT
But how does one explore such a large dataset of tables with thousands of rows? You could upload this into a spreadsheet tool or in a programming language like R, but you would still have to do a lot of manual filtering and parsing of the dataset to make it useful for downstream analyses.
In response to this the SPAAM Community have also developed a companion tool ‘AMDirT’ to facilitate this (Borry et al. 2024). Amongst other functionality, AMDirT allows you to load different releases of AncientMetagenomeDir, filter and explore to specific samples or libraries of interest, and then generate download scripts, configuration files for pipelines, and reference BibTeX files for you, both via a command-line (CLI) or graphical user interface (GUI)!
16.4.1 Running AMDirT viewer
We will now demonstrate how to use the AMDirT graphical user interface to load a dataset, filter to samples of interest, and download some configuration input files for downstream ancient DNA pipelines.

This tutorial will require a web-browser! Make sure to run on your local laptop/PC or, if on on a server, with X11 forwarding activated.
First, we will need to activate a conda environment, and then install the latest development version of the tool for you.
While in the accessing-ancient-metagenomic-data conda environment, run the following command to load the GUI into your web-browser. If the browser doesn’t automatically load, copy the IP address and paste it in your browser’s URL bar.
amdirt viewerThe first time opening AMDirT (a streamlit app), it may ask you to sign up for a newsletter using your email.
Do not type anything when prompted (i.e., just press enter), this is entirely optional and will not affect the usage of the tool.
You will not be asked again when in the same conda environment.
Your web browser should now load, and you should see a two panel page.
Under Select a table use the dropdown menu to select ‘ancientsinglegenome-hostassociated’.
You should then see a table (Figure 16.3), pretty similar what you are familiar with with spreadsheet tools such as Microsoft Excel or LibreOffice calc.
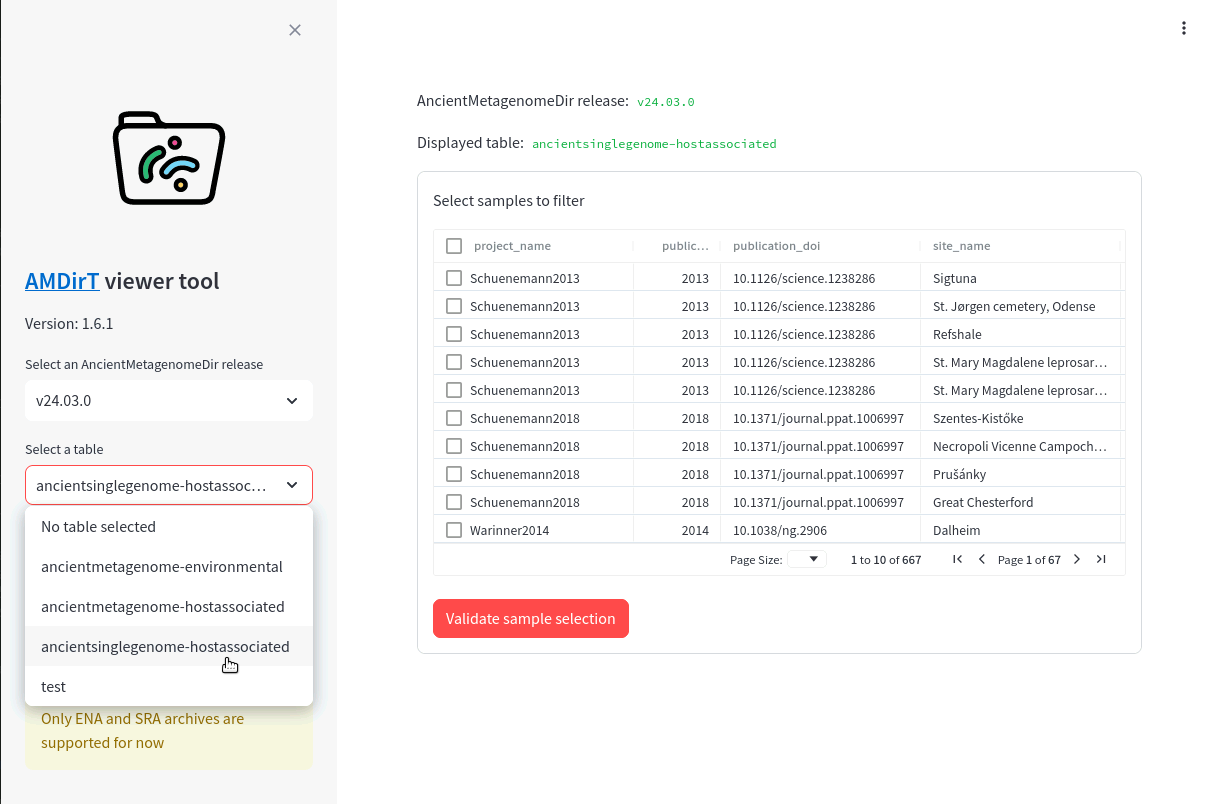
To navigate, you can scroll down to see more rows, and press shift and scroll to see more columns, or use click on a cell and use your arrow keys (⬆,⬇,⬅,➡) to move around the table.
You can reorder columns by clicking on the column name, and also filter by pressing the little ‘burger’ icon that appears on the column header when you hover over a given column.
As an exercise, we will try filtering to a particular set of samples, then generate some download scripts, and download the files.
First, filter the project_name column to ‘Muhlemann2020’ (Figure 16.4) to select data from (Mühlemann et al. 2020) .

Then scroll to the right, and filter the geo_loc_name to ‘Norway’ (Figure 16.5).

You should be left with 2 rows.
Finally, scroll back to the first column and tick the boxes of these two samples (Figure 16.6).
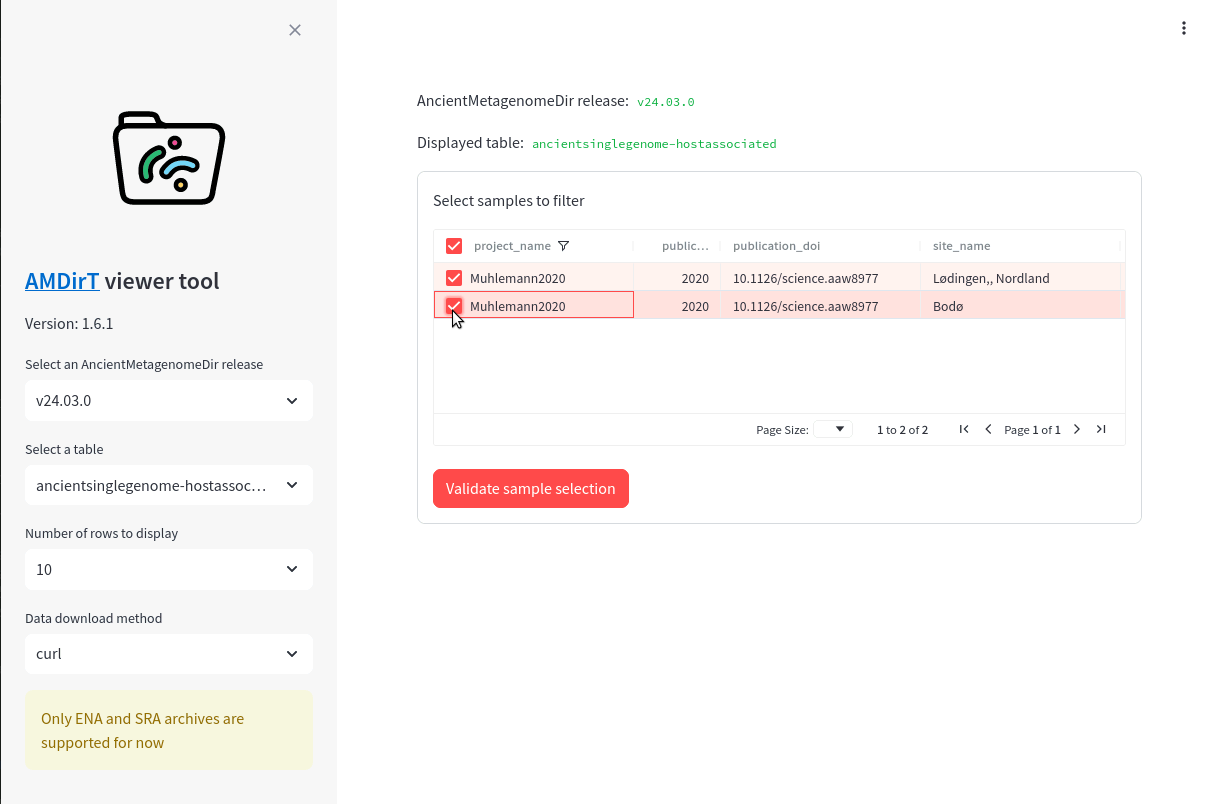
Once you’ve selected the samples you want, you can press Validate selection below the table box. You should then see a series loading-spinner, and then a new table will appear below (Figure 16.7).
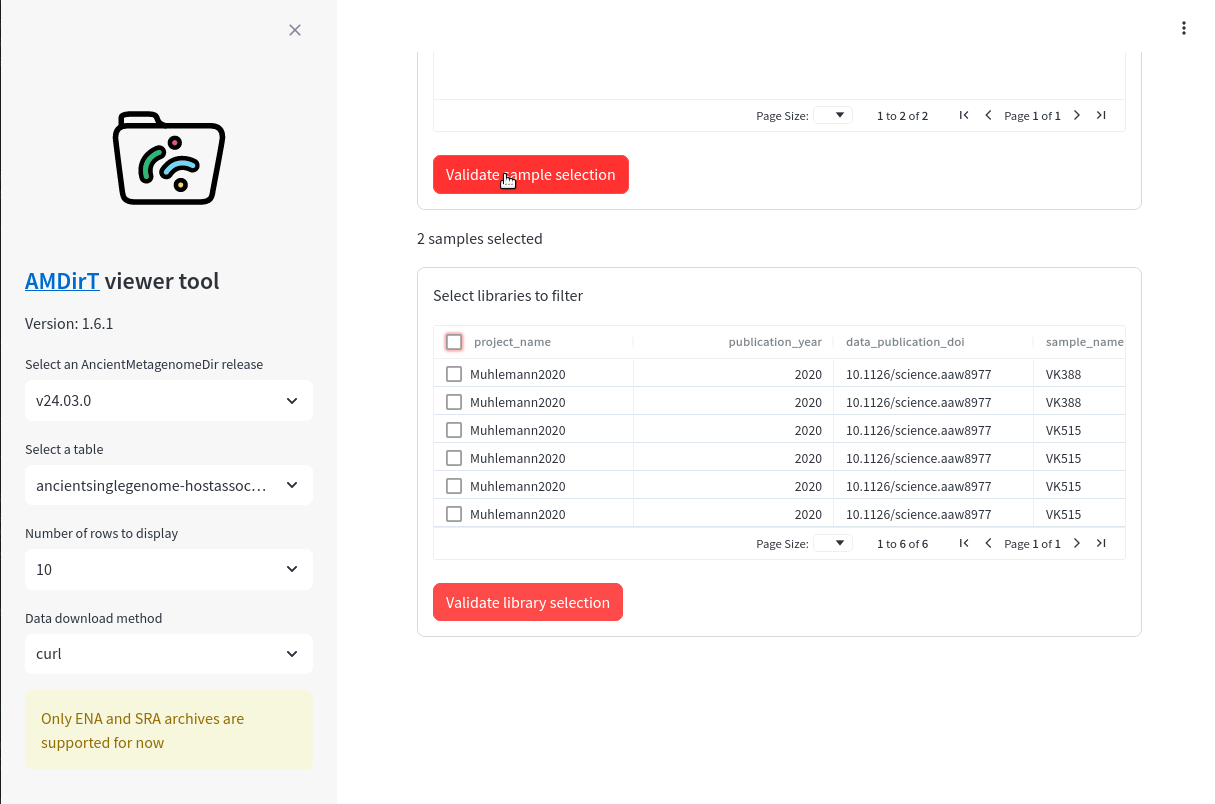
This table contains all the AncientMetagenomeDir recorded library metadata for the samples you selected.
You can filter this table in exactly the same way as you did for the samples table, and reorder columns in the same way. For example, let’s select only libraries that underwent a genome capture (Figure 16.8).

Again, you can select the libraries of interested after the filtering (Figure 16.9).

Once selected, you can press Validate library selection. A lot of buttons should then appear below the library table (Figure 16.10)!

You should have four categories of buttons:
- Download AncientMetagenomeDir Library Table
- Download Curl sample download script
- Download <tool/pipeline name> input TSV
- Download Citations as BibText
The purposes of the buttons are as follows:
- The first button is to download a table containing all the AncientMetagenomeDir metadata of the selected samples.
- The second is for generating a download script that will allow you to immediately download all sequencing data of the samples you selected.
- The third set of buttons generate (partially!) pre-configured input files for use in dedicated ancient DNA pipeline such as nf-core/eager (Fellows Yates, Lamnidis, et al. 2021), PALAEOMIX (Schubert et al. 2014), and/or and aMeta (Pochon et al. 2022).
- Finally, the fourth button generates a text file with (in most cases) all the citations of the data you downloaded, in a format accepted by most reference/citation managers.
It’s important to note you are not necessarily restricted to Curl (https://curl.se/) for downloading the data. AMDirT aims to add support for whatever tools or pipelines requested by the community. For example, an already supported downloading tool alternative is the nf-core/fetchNGS (https://nf-co.re/fetchngs) pipeline. You can select these using the drop-down menus on the left hand-side.
For the next step of this tutorial, we will press the following buttons:
- Download AncientMetagenomeDir Library Table
- Download Curl sample download script
- Download nf-core/eager input TSV
- Download Citations as BibText
Your browser should then download two .tsv files, one .sh file and one .bib file into it’s default location.
Once everything is downloaded, you can close the tab of the web browser, and in the terminal you can press ctrl + c to shutdown the tool.
Do not worry if you get a ‘Citation information could not be resolved’ warning! This is occasionally expected for some publications due to a CrossRef metadata information problem when requesting reference information about a DOI.
16.4.2 Inspecting AMDirT viewer Output
Lets look at the files that AMDirT has generated for you.
First we should mv our files from the directory that your web browser downloaded the files into into somewhere safer.
To start, we make sure we’re still in the chapter’s data directory (/<path>/<to>/accessing-ancient-metagenomic-data/), and move move the files into it.
mv ~/Downloads/AncientMetagenomeDir_* .The default download directory on most Unix desktops is normally something like ~/Downloads/, however may vary on your machine!
Then we can look inside the chapter’s directory. We should see at least the following four files.
lsAncientMetagenomeDir_bibliography.bib
AncientMetagenomeDir_curl_download_script.sh
AncientMetagenomeDir_filtered_samples.tsv
AncientMetagenomeDir_nf_core_eager_input_table.tsvWe can simply run cat of the four files downloaded by AMDirT to look inside (the files starting with AncientMetagenomeDir_). If you run cat on the curl download script, you should see a series of curl commands with the correct ENA links for you for each of the samples you wish to download.
cat AncientMetagenomeDir_curl_download_script.sh#!/usr/bin/env bash
curl -L ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR409/000/ERR4093860/ERR4093860_1.fastq.gz -o ERR4093860_1.fastq.gz
curl -L ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR409/000/ERR4093860/ERR4093860_2.fastq.gz -o ERR4093860_2.fastq.gz
curl -L ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR409/006/ERR4093846/ERR4093846.fastq.gz -o ERR4093846.fastq.gz
curl -L ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR409/005/ERR4093845/ERR4093845.fastq.gz -o ERR4093845.fastq.gz
curl -L ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR409/008/ERR4093838/ERR4093838.fastq.gz -o ERR4093838.fastq.gzBy providing this script for you, AMDirT facilitates fast download of files of interest by replacing the one-by-one download commands for each sample with a single command!
bash AncientMetagenomeDir_curl_download_script.sh % Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
0 0 0 0 0 0 0 0 --:--:-- --:--:-- --:--:-- 0
curl: (28) Timeout was reached
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
0 0 0 0 0 0 0 0 --:--:-- --:--:-- --:--:-- 0
curl: (28) Timeout was reached
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 7749k 100 7749k 0 0 12.5M 0 --:--:-- --:--:-- --:--:-- 12.6M
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 836k 100 836k 0 0 1634k 0 --:--:-- --:--:-- --:--:-- 1631k
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 4042k 100 4042k 0 0 7117k 0 --:--:-- --:--:-- --:--:-- 7117k
Running this command should result in progress logs of the downloading of the data of the four selected samples!
We can check the output by running ls to verify we have seven FASTQ files (five single end, and one paired end libraries).
Once the four samples are downloaded, AMDirT then facilitates fast processing of the data, as the eager script can be given directly to nf-core/eager as input. Importantly by including the library metadata (mentioned above), researchers can leverage the complex automated processing that nf-core/eager can perform when given such relevant metadata.
cat AncientMetagenomeDir_nf_core_eager_input_table.tsvSample_Name Library_ID Lane Colour_Chemistry SeqType Organism Strandedness UDG_Treatment R1 R2 BAM
VK388 ERR4093838 0 4 SE Homo sapiens double none ERR4093838.fastq.gz NA NA
VK515 ERR4093845 0 4 SE Homo sapiens double none ERR4093845.fastq.gz NA NA
VK515 ERR4093846 0 2 SE Homo sapiens double none ERR4093846.fastq.gz NA NA
VK515 ERR4093860 0 2 PE Homo sapiens double none ERR4093860_1.fastq.gz ERR4093860_2.fastq.gz NAFinally, we can look into the BibTeX citations file (*bib) which will provide you with the citation information of all the downloaded data and AncientMetagenomeDir itself.
The contents of this file is reliant on indexing of publications on CrossRef. In some cases not all citations will be present (as per the warning), so this should be double checked!
cat AncientMetagenomeDir_bibliography.bib @article{M_hlemann_2020, title={Diverse variola virus (smallpox) strains were widespread in northern Europe in the Viking Age}, volume={369}, ISSN={1095-9203}, url={http://dx.doi.org/10.1126/science.aaw8977}, DOI={10.1126/science.aaw8977}, number={6502}, journal={Science}, publisher={American Association for the Advancement of Science (AAAS)}, author={Mühlemann, Barbara and Vinner, Lasse and Margaryan, Ashot and Wilhelmson, Helene and de la Fuente Castro, Constanza and Allentoft, Morten E. and de Barros Damgaard, Peter and Hansen, Anders Johannes and Holtsmark Nielsen, Sofie and Strand, Lisa Mariann and Bill, Jan and Buzhilova, Alexandra and Pushkina, Tamara and Falys, Ceri and Khartanovich, Valeri and Moiseyev, Vyacheslav and Jørkov, Marie Louise Schjellerup and Østergaard Sørensen, Palle and Magnusson, Yvonne and Gustin, Ingrid and Schroeder, Hannes and Sutter, Gerd and Smith, Geoffrey L. and Drosten, Christian and Fouchier, Ron A. M. and Smith, Derek J. and Willerslev, Eske and Jones, Terry C. and Sikora, Martin}, year={2020}, month=jul }
@article{Fellows_Yates_2021, title={Community-curated and standardised metadata of published ancient metagenomic samples with AncientMetagenomeDir}, volume={8}, ISSN={2052-4463}, url={http://dx.doi.org/10.1038/s41597-021-00816-y}, DOI={10.1038/s41597-021-00816-y}, number={1}, journal={Scientific Data}, publisher={Springer Science and Business Media LLC}, author={Fellows Yates, James A. and Andrades Valtueña, Aida and Vågene, Åshild J. and Cribdon, Becky and Velsko, Irina M. and Borry, Maxime and Bravo-Lopez, Miriam J. and Fernandez-Guerra, Antonio and Green, Eleanor J. and Ramachandran, Shreya L. and Heintzman, Peter D. and Spyrou, Maria A. and Hübner, Alexander and Gancz, Abigail S. and Hider, Jessica and Allshouse, Aurora F. and Zaro, Valentina and Warinner, Christina}, year={2021}, month=jan }
This file can be easily loaded into most reference managers and then have all the citations quickly added to your manuscripts.
As in note above sometimes not all citation information can be retrieved using web APIs, so you should always validate all citations of all data you have downloaded are represented.
16.4.3 AMDirT download and convert
If you’re less of a GUI person and consider yourself a command-line wizard, you can also use the amdirt download and amdirt convert commands instead of the GUI version.
Make a new directory called cli, and change into it.
mkdir cli && cd cli/In this case you must supply your own filtered AncientMetagenomeDir samples table, and use command line options to specify which files to generate.
For this, we will download the samples table, use a bit of bash filtering, and then use the amdirt convert command to generate the same downstream-ready files as we did with the GUI.
First we can download the ancientsinglegenome-hostassociated samples table with the following command.
amdirt download -t ancientsinglegenome-hostassociated -y samplesThis will produce a file called ancientsinglegenome-hostassociated_samples_<DATA_VERSION>.tsv in your current directory (by default).
You will need to replace <DATA_VERSION> with the version number of the AncientMetagenomeDir release you are using in all commands of this tutorial!
lsancientsinglegenome-hostassociated_samples_<DATA_VERSION>.tsvThen we can use a bit of bash to filter the table in the same way as we did in the GUI. In this command, we tell awk that the column separator is a tab, print the row if either it’s the first record (line of the file), or if column one matches Muhlemann2020 and column seven matches Norway.
awk -F "\t" 'NR==1 || $1 == "Muhlemann2020" && $7 == "Norway"' ancientsinglegenome-hostassociated_samples_<DATA_VERSION>.tsv > ancientsinglegenome-hostassociated_samples_<DATA_VERSION>_filtered.tsvThen, we can pass this filtered table to the amdirt convert command to firstly retrieve the library-level metadata.
amdirt convert --librarymetadata ancientsinglegenome-hostassociated_samples_<DATA_VERSION>_filtered.tsv ancientsinglegenome-hostassociatedThis has downloaded a new file called AncientMetagenomeDir_filtered_libraries.tsv, which we can then further filter in the same away to match the desired libraries as we picked during the GUI section of the tutorial!
awk -F "\t" 'NR==1 || $14 == "Targeted-Capture"' AncientMetagenomeDir_filtered_libraries.tsv > AncientMetagenomeDir_filtered_libraries_capturedonly.tsvThen with these two filtered files, one for samples,and one for libraries, we can supply them to the convert command to generate the same download scripts, eager input samplesheet, and citation .bib file as we did before!
amdirt convert -o . --bibliography --curl --eager --libraries AncientMetagenomeDir_filtered_libraries_capturedonly.tsv ancientsinglegenome-hostassociated_samples_<DATA_VERSION>_filtered.tsv ancientsinglegenome-hostassociatedYou should see a few messages saying Writing <XYZ>, and then if we run ls, you should see the same resulting files starting with AncientMetagenomeDir_ as before with the GUI!
lsAncientMetagenomeDir_bibliography.bib
AncientMetagenomeDir_curl_download_script.sh
AncientMetagenomeDir_filtered_libraries.tsv
AncientMetagenomeDir_filtered_libraries_capturedonly.tsv
AncientMetagenomeDir_nf_core_eager_input_table.tsv
ancientsinglegenome-hostassociated_samples_<DATA_VERSION>.tsv
ancientsinglegenome-hostassociated_samples_<DATA_VERSION>_filtered.tsvTherefore the convert command route of AMDirT allows you to include AMDirT in more programmatic workflows to make downloading data more efficiently.
16.4.4 AMDirT Practise
Try to use your preferred AMDirT interface (GUI or CLI) to find the number of all single-stranded libraries of palaeofaeces metagenomes, published since 2021
The answer is 19 libraries across 1 publications: Maixner 2021 (2021)
You can calculate this with the CLI method as follows.
## Download the ancient metagenome table
amdirt download -t ancientmetagenome-hostassociated -y samples
## Filter to just palaeofaeces since 2021
awk -F "\t" 'NR==1 || $2 >= 2021 && $13 == "palaeofaeces"' ancientmetagenome-hostassociated_samples_<DATA_VERSION>.tsv > ancientmetagenome-hostassociated_samples_<DATA_VERSION>_filtered.tsv
## Get the library metadata of the dental calculus (might take a little bit of time)
amdirt convert --librarymetadata ancientmetagenome-hostassociated_samples_<DATA_VERSION>_filtered.tsv ancientmetagenome-hostassociated
## Filter to just single stranded libraries
awk -F "\t" 'NR==1 || $9 == "single"' AncientMetagenomeDir_filtered_libraries.tsv > AncientMetagenomeDir_filtered_libraries_singlestranded.tsv
## Count the number of libraries, starting from row 2 to skip the header
tail -n +2 AncientMetagenomeDir_filtered_libraries_singlestranded.tsv | wc -l16.5 Git Practise
A critical factor of AncientMetagenomeDir is that it is community-based. The community curates all new submissions to the repository, and this all occurs with Git and GitHub.
The data is hosted and maintained on GitHub - new publications are evaluated on issues, submissions created on branches, made by pull requests, and PRs reviewed by other members of the community.
You can see the workflow in the image below (Figure 16.11) from the original AncientMetagenomeDir publication (Fellows Yates, Andrades Valtueña, et al. 2021), and read more about the workflow on the AncientMetagenomeDir wiki (https://github.com/SPAAM-community/AncientMetagenomeDir/wiki).
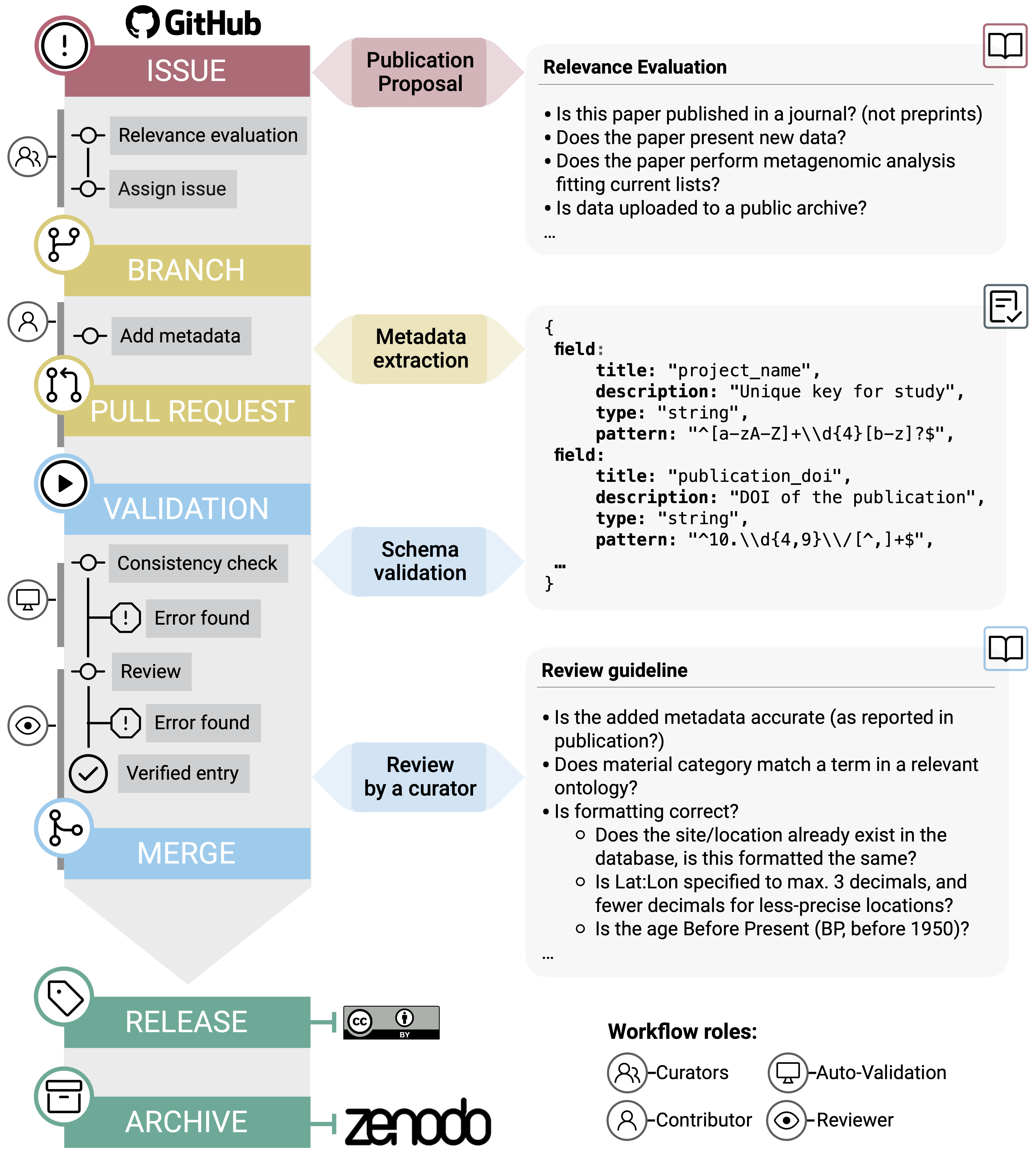
As AncientMetagenomeDir is on GitHub, the means we can also use this repository to try out our Git skills we learnt in the chapter Introduction to Git(Hub)!
Your task is to complete the following steps. However, we’ve replaced the correct Git terminology with generic verbs, indicated with words in quotes. Recreate the following, but also note down what the correct Git terminology is.
- Make a ‘copy’ the
jfy133/AncientMetagenomeDir(https://github.com/jfy133/AncientMetagenomeDir) GitHub repository to your account
We are forking a personal fork of the main repository to ensure we don’t accidentally edit the main AncientMetagenomeDir repository!
‘Download’ the copied repo to your local machine
‘Change’ into a new branch called
devModify the file
ancientsinglegenome-hostassociated_samples.tsv. Add the following line to the end of the TSVMake sure your text editor doesn’t replace tabs with spaces!
Long2022 2022 10.1038/s42003-022-03527-1 Basilica of St. Domenico Maggiore 40.848 14.254 Italy NASD1 Homo sapiens 400 10.1038/s42003-022-03527-1 bacteria Escherichia coli calcified nodule chromosome SRA raw PRJNA810725 SRS12115743‘Save’, ‘Record’, and ‘Send’ back to Git(Hub)
Open a ‘merge suggestion’ proposing the changes to the original
jfy133/AncientMetagenomeDirrepo- Make sure to put ‘Summer school’ in the title of the ‘Request’
- Fork the
jfy133/AncientMetagenomeDir(https://github.com/jfy133/AncientMetagenomeDir) repository to your account (Figure 16.12)
Clone the copied repo to your local machine
git clone git@github.com:<YOUR_USERNAME>/AncientMetagenomeDir.Git cd AncientMetagenomeDirSwitch to a new branch called
devgit switch -c devModify
ancientsinglegenome-hostassociated_samples.tsvecho "Long2022 2022 10.1038/s42003-022-03527-1 Basilica of St. Domenico Maggiore 40.848 14.254 Italy NASD1 Homo sapiens 400 10.1038/s42003-022-03527-1 bacteria Escherichia coli calcified nodule chromosome SRA raw PRJNA810725 SRS12115743" >> ancientsinglegenome-hostassociated/samples/ancientsinglegenome-hostassociated_samples.tsvAdd, Commit and Push back to your Fork on Git(Hub)
git add ancientsinglegenome-hostassociated/samples/ancientsinglegenome-hostassociated_samples.tsv git commit -m 'Add Long2022' git pushOpen a Pull Request adding changes to the original jfy133/AncientMetagenomeDir repo (Figure 16.13)
- Make sure to make the pull request against
jfy133/AncientMetagenomeDirand NOTSPAAM-community/AncientMetagenomeDir - Make sure to put ‘Summer school’ in the title of the pull request!
- Make sure to make the pull request against
16.6 Summary
- Reporting of metadata messy! Consider when publishing your own work!
- Use AncientMetagenomeDir as a template for supplementary tables!
- Use AncientMetagenomeDir and AMDirT to rapidly find and download public ancient metagenomic data
- You can use it to generate templates for dowsntream processing pipelines!
- Contribute to AncientMetagenomeDir with git
- It is community curated: it will be as good as you make it, the more people who contribute, the easier and better it is.
16.7 (Optional) clean-up
Let’s clean up your working directory by removing all the data and output from this chapter.
When closing your jupyter notebook(s), say no to saving any additional files.
Press ctrl + c on your terminal, and type y when requested. Once completed, the command below will remove the /<PATH>/<TO>/accessing-metagenomic-data directory as well as all of its contents.
Always be VERY careful when using rm -r. Check 3x that the path you are specifying is exactly what you want to delete and nothing more before pressing ENTER!
rm -r /<PATH>/<TO>/accessing-ancient-metagenomic-data*Once deleted you can move elsewhere (e.g. cd ~).
We can also get out of the conda environment with
conda deactivateTo delete the conda environment
conda remove --name accessing-ancient-metagenomic-data --all -y