17 Ancient Metagenomic Pipelines
For this chapter’s exercises, if not already performed, we will need to download the chapter’s dataset, decompress the archive, and create and activate the conda environment.
Do this, use wget or right click and save to download this Zenodo archive: 10.5281/zenodo.17155341, and unpack.
tar xvf ancient-metagenomic-pipelines.tar.gz
cd ancient-metagenomic-pipelines/We can then create the subsequently activate environment with the following commands.
conda env create -f ancient-metagenomic-pipelines.yml
conda activate ancient-metagenomic-pipelinesThere are additional software requirements for this chapter
Please see the relevant chapter section in Before we start before continuing with this chapter.
17.1 Introduction
A pipeline is a series of linked computational steps, where the output of one process becomes the input of the next. Pipelines are critical for managing the huge quantities of data that are now being generated regularly as part of ancient DNA analyses. In this chapter we will go through three dedicated ancient DNA pipelines - all with some (or all!) functionality geared to ancient metagenomics - to show we how we can speed up the more routine aspects of the basic analyses we’ve learnt about earlier in this text book through workflow automation.
We will introduce:
- nf-core/eager (https://nf-co.re/eager) - a generalised aDNA ‘workhorse’ pipeline that can do both ancient genomics and (basic) metagenomics (Fellows Yates et al. 2021)
- aMeta (https://github.com/NBISweden/aMeta) - a pipeline for resource efficient and accurate ancient microbial detection and authentication (Pochon et al. 2022)
- nf-core/mag (https://nf-co.re/mag) - a de novo metagenomics assembly pipeline (Krakau et al. 2022) that includes a dedicated ancient DNA mode for damage correction and validation.
Keep in mind that that there are many other pipelines that exist, and picking which one often they come down to personal preference, such as which functionality they support, which language they are written in, and whether their computational requirements can fit in our available resources.
Other examples of other ancient DNA genomic pipelines include Paleomix (https://paleomix.readthedocs.io/en/stable, Schubert et al. 2014), and Mapache (https://github.com/sneuensc/mapache, Neuenschwander et al. 2023), and for ancient metagenomics: metaBit (https://bitbucket.org/Glouvel/metabit/src/master/, Louvel et al. 2016) and HAYSTAC (https://github.com/antonisdim/haystac, Dimopoulos et al. 2022).
17.2 Workflow managers
All the pipelines introduced in this chapter utilise workflow managers. These are software that allows users to ‘chain’ together the inputs and outputs distinct ‘atomic’ steps of a bioinformatics analysis - e.g., separate terminal commands of different bioinformatic tools, so that ‘we don’t have to’. we have already seen very basic workflows or ‘pipelines’ when using the bash ‘pipe’ (|) in the Introduction to the Command Line chapter, where the each row of text the output of one command was ‘streamed’ into the next command.
However in the case of bioinformatics, we are often dealing with non-row based text files, meaning that ‘classic’ command line pipelines don’t really work. Instead this is where bioinformatic workflow managers come in: they handle the passing of files from one tool to the next but in a reproducible manager.
Modern computational bioinformatic workflow managers focus on a few main concepts. To summarise Wratten et al. (2021), these areas are: data provenance, portability, scalability, re-entrancy - all together which contribute to ensuring reproducibility of bioinformatic analyses.
Data provenance refers to the ability to track and visualise where each file goes and gets processed, as well as metadata about each file and process (e.g., What version of a tool was used? What parameters were used in that step? How much computing resources were used).
Portability follows from data provenance where it’s not just can the entire execution of the pipeline be reconstructed - but can it also be run with the same results on a different machine? This is important to ensure that we can install and test the pipeline on our laptop, but when we then need to do heavy computation using real data, that it will still be able to execute on a high-performance computing cluster (HPC) or on the cloud - both that have very different configurations. This is normally achieved through the use of reusable software environments such as as conda (https://docs.conda.io/en/latest/) or container engines such as docker (https://www.docker.com/), and tight integration with HPC schedulers such as SLURM (https://slurm.schedmd.com/documentation.html).
As mentioned earlier, not having to run each command manually can be a great speed up to our analysis. However this needs to be able to be Scalable that it the workflow is still efficient regardless whether we’re running with one or ten thousands samples - modern workflow managers perform resource requirement optimisation and scheduling to ensure that all steps of the pipeline will be executed in the most resource efficient manner so it completes as fast as possible - but regardless of of the number of input data.
Finally, as workflows get bigger and longer, re-entrancy has become more important, i.e., the ability to re-start a pipeline run that got stuck halfway through due to an error.
All workflow managers have different ways of implementing the concepts above, and these can be very simple (e.g., Makefiles, https://en.wikipedia.org/wiki/Make_(software))) to very powerful and abstract (e.g. Workflow Description Language, https://github.com/openwdl/wdl). In this chapter we will use pipelines that use two popular workflow managers in bioinformatics, Nextflow (https://nextflow.io, (Di Tommaso et al. 2017)) and Snakemake (https://snakemake.github.io, (Mölder et al. 2021)).
This chapter will not cover how to write our own workflow, as this would require a whole other textbook. However it is recommended to learn and use workflow managers when carrying out repetitive or routine bioinformatic analysis (Nextflow and Snakemake being two popular ones in bioinformatics). Use of workflow managers can help make our work more efficiently (as we only run one command, rather than each step separately), but also more reproducible by reducing the risk of user error when executing each step: the computer will do exactly what we tell it, and if we don’t change anything, will do the exact same thing every time. If you’re interested in writing our own workflows using workflow managers, many training and tutorials exist on the internet (e.g., for Nextflow there is the official training: https://training.nextflow.io/ or from software carpentries: https://carpentries-incubator.github.io/workflows-nextflow/, or the official training for snakemake: https://snakemake.readthedocs.io/en/stable/tutorial/tutorial.html#tutorial).
17.3 What is nf-core/eager?
nf-core/eager is a computational pipeline specifically designed for preprocessing and analysis of ancient DNA data (Figure 17.1) (Fellows Yates et al. 2021). It is a reimplementation of the previously published EAGER (Efficient Ancient Genome Reconstruction) pipeline (Peltzer et al. 2016) written in the workflow manager Nextflow. In addition to reimplementing the original genome mapping and variant calling pipeline, in a more reproducible and portable manner the pipeline also included additional new functionality particularly for researchers interested in microbial sciences, namely a dedicated genotyper and consensus caller designed for low coverage genomes, the ability to get breadth and depth coverage statistics for particular genomic features (e.g. virulence genes), but also automated metagenomic screening and authentication of the off-target reads from mapping (e.g. against the host reference genome).
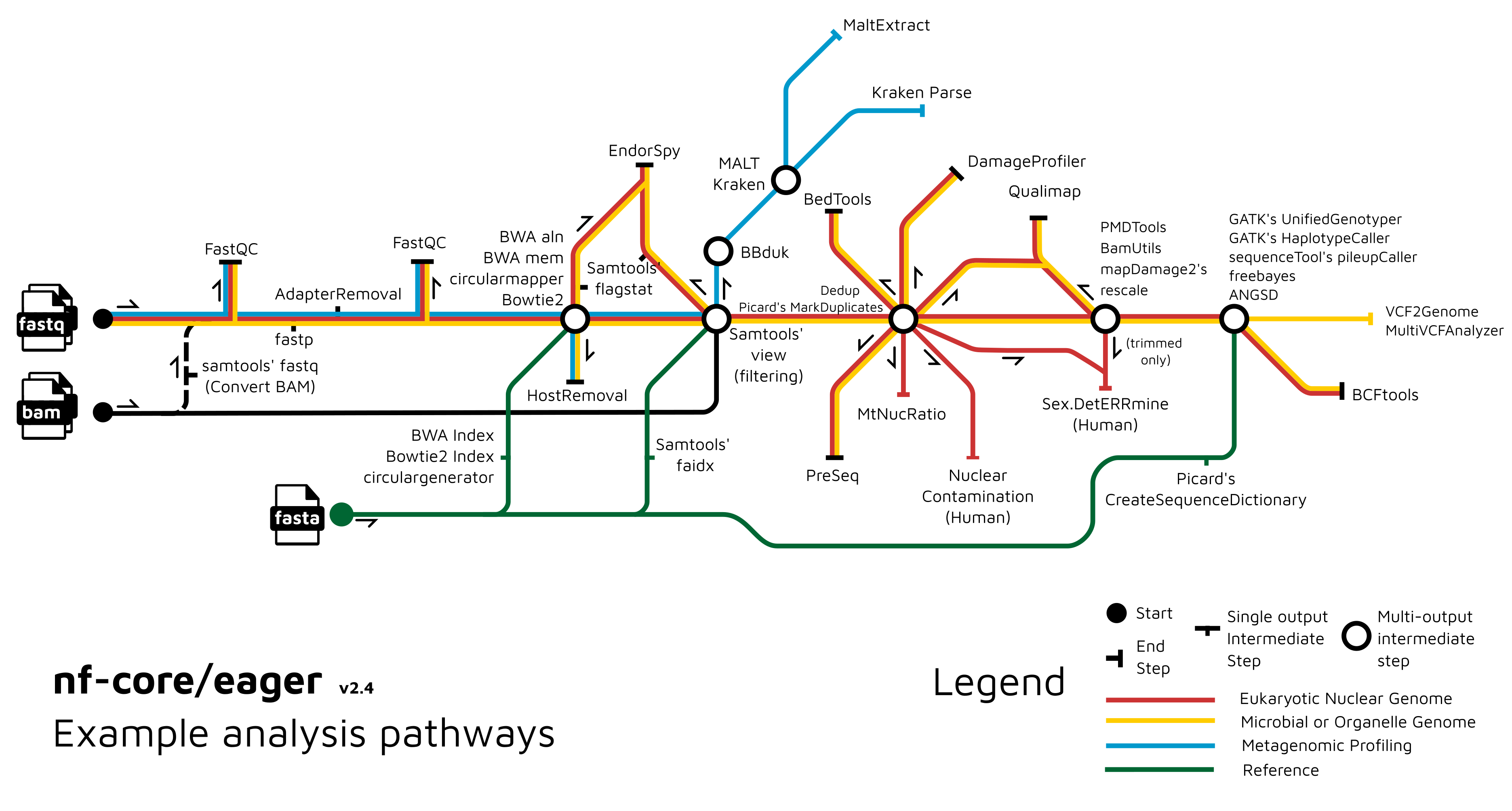
FastQC, AdapterRemoval, alignment to a reference FASTA input with a range of alignments, before going through BAM filtering, deduplication, in silico damage removal, and variant calling. Multiple statistics steps come out of the deduplication ‘station’. The blue line represents the metagenomic workflow, where off-target reads come out of the BAM filtering ‘station’, and goes through complexity filtering with BBDuk, MALT or Kraken2, and optionally into MaltExtract for MALT output.
A detailed description of steps in the pipeline is available as part of nf-core/eager’s extensive documentation. For more information, check out the usage documentation (https://nf-co.re/eager/2.5.2/docs/usage/).
Briefly, nf-core/eager takes at a minimum standard input file types that are shared across the genomics field, i.e., raw FASTQ files or aligned reads in bam format, and a reference fasta. nf-core/eager performs preprocessing of this raw data, including adapter clipping, read merging, and quality control of adapter-trimmed data. nf-core/eager then carries mapping using a variety of field-standard shot-read alignment tools with default parameters adapted for short and damaged aDNA sequences. The off-target reads from host DNA mapping can then go into metagenomic classification and authentication (in the case of MALT (Vågene et al. 2018; Herbig et al. 2016)). After genomic mapping, BAM files go through deduplication of PCR duplicates, damage profiling and removal, and finally variant calling. A myriad of additional statistics can be generated depending on the users preference. Finally, nf-core eager uses MultiQC (https://multiqc.info/, (Ewels et al. 2016)) to create an integrated html report that summarises the output/results from each of the pipeline steps.
Why cannot we use a standard genomics mapping and variant calling pipeline, such as nf-core/sarek Hanssen et al. (2024) for ancient DNA ?
Many tools used in standard genomics pipelines assume ‘modern’ data quality, i.e., high coverage, low error rates, and long read lengths.
In ancient DNA we need to use tools better suited for low coverage, and short read lengths. Furthermore, we would like additional tools for authenticating our ancient DNA - characteristics that you would not expect to find in modern data and thus not included in modern pipelines.
17.3.1 Running nf-core/eager
For the practical portion of this chapter, we will utilise sequencing data from four aDNA libraries, which we should have already downloaded from NCBI. If not, please see the Preparation section above. We will use nf-core/eager to perform a typical microbial genomic analysis, i.e., reconstruction of an ancient genome to generate variant calls that can be used for generating phylogenomic trees and other evolutionary analysis, and gene feature coverage statistics to allow insight into the functional aspects of the genome.
These four libraries come from from two ancient individuals, GLZ002 and KZL002. GLZ002 comes from the Neolithic Siberian site of Glazkovskoe predmestie (Yu et al. 2020) and was radiocarbon dated to 3081-2913 calBCE. KZL002 is an Iron Age individual from Kazakhstan, radiocarbon dated to 2736-2457 calBCE (Andrades Valtueña et al. 2022). Both individuals were originally analysed for human population genetic analysis, but when undergoing metagenomic screening of the off-target reads, both set of authors identified reads from Yersinia pestis from these individuals - the bacterium that causes plague. Subsequently the libraries from these individuals were processed using hybridisation capture to increase the number of Y. pestis sequences available for analysis.
Our aims in the following tutorial are to:
- Preprocess the FASTQ files by trimming adapters and merging paired-end reads
- Align reads to the Y. pestis reference and compute the endogenous DNA percentage
- Filter the aligned reads to remove host DNA
- Remove duplicate reads for accurate coverage estimation and genotyping
- Generate statistics on gene features (e.g. virulence factors)
- Merge data by sample and perform genotyping on the combined dataset
- Review quality control data to evaluate the success of the previous steps
Let’s get started!
nf-core/eager v2 requires an older version of Nextflow - if installing manually, ensure we do not have Nextflow version any greater than v22.10.6!
First, download the latest version of the nf-core/eager repo (or check for updates if we have a previously-installed version).
nextflow pull nf-core/eagerNext we can re-visit AMDirT (see Accessing Ancient Metagenomic Data, (Borry et al. 2024)) to download a pre-prepared configuration file for nf-core/eager
- Load AMDirt (
amdirt viewer), and select the latest release and the ‘ancientsinglegenome-hostassociated’ table - Filter the
sample_namecolumn to just show KZL002 and GLZ002, and select these two rows- Press the burger icon on the column
- Press the filter tab and deselect everything
- Search GLZ002 and select in filter menu
- Search KZL002 and select in filter menu (careful: KZL not KLZ!)
- Close filter menu and select the two rows
- Press the Validate Selection button
- Press the ‘Download Curl sample download script’, ‘Download nf-core/eager input TSV’, and ‘Download Citations as BibTex’ buttons
- Move the downloaded files into
eager/of this tutorial’s directory- i.e., if the files were downloaded to we
Downloads/folder rather than the chapters directory, - e.g., assuming we’ve got no other previous downloads we can run
mv ~/Downloads/AncientMetagenomeDir_* eager/
- i.e., if the files were downloaded to we
- Close AMDiRT in the web browser and shut it down in the terminal with
ctrl + c
We won’t actually use the BibTex citations file for anything in this tutorial, but it is good habit to always to record and save all citations of any software or data we use!
To download the FASTQ files
- Move into the
eager/directory - Run
bash AncientMetagenomeDir_curl_download_script.shto download the files (this may take ~3 minutes)
Next we now inspect the AMDirT generated input TSV file for nf-core/eager!
cat AncientMetagenomeDir_nf_core_eager_input_table.tsvSample_Name Library_ID Lane Colour_Chemistry SeqType Organism Strandedness UDG_Treatment R1 R2 BAM
GLZ002 ERR4093961 0 4 PE Homo sapiens double half ERR4093961_1.fastq.gz ERR4093961_2.fastq.gz NA
GLZ002 ERR4093962 0 4 PE Homo sapiens double full ERR4093962_1.fastq.gz ERR4093962_2.fastq.gz NA
KZL002 ERR8958768 0 4 PE Homo sapiens double half ERR8958768_1.fastq.gz ERR8958768_2.fastq.gz NA
KZL002 ERR8958769 0 4 PE Homo sapiens double half ERR8958769_1.fastq.gz ERR8958769_2.fastq.gz NAHere we see 10 columns, all pre-filled. The first two columns correspond to sample/library IDs that will be used for data provenance and grouping. When we have sequencing multiple lanes we can speed up preprocessing these independently before merging, so lane can specify this (although not used in this case as we have independent libraries per sample. we can indicate the colour chemistry to indicate whether our data requires additional pre-processing to remove poly-G tails, and then also strandedness and UDG damage treatment status of the libraries if we require further damage manipulation. Finally we provide paths to the FASTQ files or BAM files.
Other than the raw FASTQ files, we will need a reference genome and annotation coordinates of genes present on the genome. In this case we will use a Yersinia pestis (accession: GCF_001293415.1) reference genome (.fna) and gene feature file (.gff) from NCBI Genome (https://www.ncbi.nlm.nih.gov/genome).
These have already been placed in the reference/ directory for us.
To download the required reference genome and annotation file run the following command to download from the NCBI Genome database.
## Download from NCBI
curl -OJX GET "https://api.ncbi.nlm.nih.gov/datasets/v2alpha/genome/accession/GCF_001293415.1/download?include_annotation_type=GENOME_FASTA,GENOME_GFF,RNA_FASTA,CDS_FASTA,PROT_FASTA,SEQUENCE_REPORT&filename=GCF_001293415.1.zip" -H "Accept: application/zip"
unzip *.zip
mv ncbi_dataset/data/GCF_001293415.1/* .
## We have to sort the gff file to make it eager compatible
gffread genomic.gff GCF_001293415.1_ASM129341v1_genomic.gffWith all of these, we can run the pipeline!
First lets enter a screen session to make sure we can leave the pipeline running in the background and continue using our terminal.
screen -R eager
conda activate ancient-metagenomic-pipelinesNow we can construct an eager command from within the data/ directory so that it looks like this.
nextflow run nf-core/eager -r 2.5.3 \
-profile docker \
--fasta ../reference/GCF_001293415.1_ASM129341v1_genomic.fna \
--input AncientMetagenomeDir_nf_core_eager_input_table.tsv \
--anno_file ../reference/GCF_001293415.1_ASM129341v1_genomic.gff \
--run_bam_filtering --bam_unmapped_type discard \
--skip_preseq \
--run_genotyping --genotyping_tool ug --gatk_ug_out_mode EMIT_ALL_SITES \
--run_bcftools_stats --run_bedtools_coverageWhen the run starts, we see a long list of process lines with progress bars slowly appearing over time. If we press ctrl + a and then [ to access a ‘navigation’ (called ‘copy’) mode, then use our arrow keys on our keyboard to scroll up and down. To quit this mode just press q.
We don’t normally recommend running analyses in the directory our data is in! It is better to keep data and analysis results in separate directories. Here we are just running eager alongside the data for convenience (i.e., we don’t have to modify the downloaded input TSV)
This run will take between 20m-40m to run on a standard laptop! Time for a break, or we can continue reading this chapter as the critical output data has already been provided for we in the data directory.
So what is this command doing? The different parameters do the following:
- Tell
nextflowto run the nf-core/eager pipeline with version 2.4.7 - Specify which computing and software environment to use with
-profile- In this case we are running locally so we don’t specify a computing environment (such as a configuration for an institutional HPC)
- We use
dockeras our container engine, which downloads all the software and specific versions needed for nf-core/eager in immutable ‘containers’, to ensure nothing get broken and is as nf-core/eager expects
- Provide the various paths to the input files (TSV with paths to FASTQ files, a reference fasta, and the reference fasta’s annotations)
- Activate the various of the steps of the pipeline we’re interested in
- We turn off preseq (e.g. when we know we can’t sequence more)
- We want to turn on BAM filtering, and specify to generate unmapped reads in FASTQ file (so we could check off target reads e.g. for other pathogens)
- we turn on genotyping using
GATK UnifiedGenotyper(preferred overGATK HaplotypeCaller(Poplin et al. 2018) due to in compatibility with that method to low-coverage data) - We turn on variant statistics (from GATK) using
bcftools(Danecek et al. 2021), and coverage statistics of gene features using bedtools (Quinlan and Hall 2010)
For full parameter documentation, see the website (https://nf-co.re/eager/2.5.2/parameters).
And now we wait…
As a reminder, to detach from the screen session type ctrl + a then d. To reattach screen -r eager
Specifically for ancient (meta)genomic data, the following parameters and options may be useful to consider when running our own data:
--mapper circularmapper- This aligner is a variant of
bwa alnthat allows even mapping across the end of linear references of circular genomes - Allows reads spanning the start/end of the sequence to be correctly placed, and this provides better coverage estimates across the entire genome
- This aligner is a variant of
--hostremoval_input_fastq:- Allows re-generation of input FASTQ files but with any reads aligned to the reference genome removed
- Can be useful when dealing with modern or ancient data where there are ethical restrictions on the publication of host DNA
- The output can be used as ‘raw data’ upload to public data repositories
--run_bam_filtering --bam_unmapped_type- A pre-requisite for performing the metagenomic analysis steps of nf-core/eager
- Generates FASTQ files off the unmapped reads present in the reference mapping BAM file
--run_bedtools_coverage --anno_file /<path>/<to>/<genefeature>.bed- Turns on calculating depth/breadth of annotations in the provided bed or GFF file, useful for generating e.g. virulence gene presence/absence plots
--run_genotyping --genotyping_tool ug --gatk_ug_out_mode EMIT_ALL_SITES- Turns on genotyping with
GATK UnifiedGenotyper - A pre-requisite for running
MultiVCFAnalyzer(Bos et al. 2014) for consensus sequencing creation - It’s recommend to use either
GATK UnifiedGenotyperorfreeBayes(non-MultiVCFAnalyzer compatible!, Garrison and Marth 2012) for low-coverage data GATK HaplotypeCalleris not recommended for low coverage data as it performs local de novo assembly around possible variant sites, and this will fail with low-coverage short-read data
- Turns on genotyping with
--run_multivcfanalyzer --write_allele_frequencies- Turns on SNP table and FASTA consensus sequence generation with
MultiVCFAnalyzer(see pre-requisites above) - By providing
--write_allele_frequencies, the SNP table will also provide the percentage of reads at that position supporting the call. This can help we evaluate the level of cross-mapping from related (e.g. contaminating environmental) species and may question the reliability of any resulting downstream analyses.
- Turns on SNP table and FASTA consensus sequence generation with
--metagenomic_complexity_filtering- An additional preprocessing step of raw reads before going into metagenomic screening to remove low-complexity reads (e.g. mono- or di-nucleotide repeats)
- Removing these will speed up and lower computational requirements during classification, and will not bias profiles as these sequences provide no taxon-specific information (i.e., can be aligned against thousands to millions of genomes)
--run_metagenomic_screening- Turns on metagenomic screening with either
MALTorKraken2 - If running with
MALT, can supply--run_maltextractto get authentication statistics and plots (damage, read lengths etc.) for evaluation
- Turns on metagenomic screening with either
Why is it critical to report versions of all pipelines and tools?
(Bioinformatics) software is rarely stable.
Many tools will update frequently either with bug fixes and new or optimised functionality.
Both can cause changes in the output of the software, and thus the results of our analyses. If we want our analyses to be completely reproducible, and increase trust of other scientists in our results, we need to make sure they can generate the same output as we did.
17.3.2 Top Tips for nf-core/eager success
Screen sessions
Depending on the size of our input data, infrastructure, and analyses, running nf-core/eager can take hours or even days (e.g. the example command above will likely take around 20 minutes!).
To avoid crashes due to loss of power or network connectivity, try running nf-core/eager in a
screenortmuxsession.screen -R eagerMultiple ways to supply input data
In this tutorial, a tsv file to specify our input data files and formats. This is a powerful approach that allows nf-core eager to intelligently apply analyses to certain files only (e.g. merging for paired-end but not single-end libraries). However inputs can also be specified using wildcards, which can be useful for fast analyses with simple input data types (e.g. same sequencing configuration, file location, etc.).
See the online nf-core/eager documentation (https://nf-co.re/eager/usage) for more details.
nextflow run nf-core/eager -r 2.4.7 -profile docker --fasta ../reference/GCF_001293415.1_ASM129341v1_genomic.fna \
--input "data/*_{1,2}.fastq.gz" <...> \
--udg_type halfGet our
MultiQCreport via emailIf we have
GNU mailorsendmailset up on our system, we can add the following flag to send theMultiQChtml to our email upon run completion:--email "your_address@something.com"Check out the nf-core/eager launch GUI
For researchers who might be less comfortable with the command line, check out the nf-core/eager launch GUI (https://nf-co.re/launch?pipeline=eager&release=2.5.2)! The GUI also provides a full list of all pipeline options with short explanations for those interested in learning more about what the pipeline can do.
When something fails, all is not lost!
When individual jobs fail, nf-core/eager will try to automatically resubmit that job with increased memory and CPUs (up to two times per job).
When the whole pipeline crashes, we can save time and computational resources by resubmitting with the
-resumeflag. nf-core/eager will retrieve cached results from previous steps as long as the input is the same.Monitor our pipeline in real time with the Seqera Platform
Regular users may be interested in checking out the Seqera Platform (previously known as Nextflow Tower), a tool for monitoring the progress of
Nextflowpipelines in real time. Check the website (https://tower.nf) for more information.
17.3.3 nf-core/eager output: results files
Once completed, the results/ directory of our nf-core/eager run will contain a range of directories that will have output files and tool logs of all the steps of the pipeline. nf-core/eager tries to only save the ‘useful’ files.
Everyday useful files for ancient (microbial) (meta)genomics typically are in folders such as:
deduplication/ormerged_bams/- These contain the most basic BAM files we would want to use for downstream analyses (and used in the rest of the genomic workflow of the pipeline)
- They contain deduplicated BAM files (i.e., with PCR artefacts removed)
merged_bams/- This directory will contain BAMs where multiple libraries of one sample have been merged into one final BAM file, when these have been supplied
damage_rescaling/ortrimmed bam/- These contain the output BAM files from in silico damage removal, if we have turned on e.g. BAM trimming or damage rescaling
- If we have multiple libraries of one sample, the final BAMs we want will be in
merged_bams/
genotyping/- This directory will contain our final VCF files from the genotyping step, e.g. from the GATK or
freeBayestools
- This directory will contain our final VCF files from the genotyping step, e.g. from the GATK or
MultiVCFAnalyzer/- This will contain consensus sequences and SNP tables from
MultiVCFAnalyzer, which also allows generation of ‘multi-allelic’ SNP position statistics (useful for the evaluation of cross-mapping from contaminants or relatives)
- This will contain consensus sequences and SNP tables from
bedtools/- This directory will contain the depth and breadth statistics of genomic features if a
gfforbedfile has been provided to the pipeline - The files can be used to generate gene heatmaps, that can be used to visualise a comparative presence/absence of virulence factor across genomes (e.g. for microbial pathogens)
- This directory will contain the depth and breadth statistics of genomic features if a
metagenomic_classificationormalt_extract- This directory contains the output RMA6 files from
MALT, the profiles and taxon count tables fromKraken2, or the aDNA authentication output statistics frommaltExtract.
- This directory contains the output RMA6 files from
Most other folders contain either intermediate files that are only useful for technical evaluation in the case of problems, or statistics files that are otherwise summarised in the run report.
So, before we delve into these folders, it’s normally a good idea to do a ‘quality check’ of the pipeline run. we can do this using the interactive MultiQC pipeline run report.
Why is it important to use deduplicated BAMs for downstream genomic analyses?
Without deduplication, you are artificially increasing the confidence in variant calls during genotyping. Many downstream analyses assume that each base call is made up of multiple indepndent ‘observations’ of that particular nucleotide. If you have not deduplicated your alignments, you may have the exact sample molecule represented twice (an artefact of amplification), thus violating the ‘independent observation’ assumption.
17.3.4 nf-core/eager output: run report
cd multiqc/If we’re impatient, and our nf-core/eager run hasn’t finished yet, we can cancel the run with ctrl + c (possibly a couple of times), and we can open a pre-made file in the data directory under ancient-metagenomic-pipelines/multiqc_report.html
In here we should see a bunch of files, but we should open the multiqc_report.html file in our browser. We can either do this via the command-line (e.g. for firefox firefox multiqc_report.html) or navigate to the file using our file browser and double clicking on the HTML file.
During the summer school if you cannot open the MultiQC HTML file directly in the VM, copy the file to your Downloads folder and open from there.e.g.
For example the following two commands.
cp multiqc_report.html ~/Downloads/
firefox ~/Downloads/multiqc_report.htmlOnce opened we will see a table, and below it many figures and other tables (Figure 17.2). All of these statistics can help we evaluate the quality of our data, pipeline run, and also possibly some initial results!

MultiQC report. The left hand sidebar provides links to various sections of the report, most of them containing summary images of the outputs of all the different tools. On the main panel we see the MultiQC and nf-core/eager logos, some descriptive information about how the report was generated, and finally the top of the ‘General Statistics’ tables, that has columns such as Sample Name, Nr. Input reads, length of input reads, %trimmed, etc. Each numeric column contains a coloured bar chart to help readers to quickly evaluate the number of a given sample against all others in the table
Typically we will look at the General Statistics table to get a rough overview of the pipeline run.
If we hover our cursor over the column headers, we can see which tool the column’s numbers came from, however generally the columns go in order of left to right, where the left most columns are from earlier in the pipeline run (e.g. removing adapters), to variant calling statistics (e.g. number of variants called). we can also configure which columns to display using the Configure columns button.
It’s important to note that in MultiQC tables, we may have duplicate rows from the same library or sample. This is due to MultiQC trying to squish in as many statistics from as many steps of the pipeline as possible (for example, statistics on each of a pair of FASTQ files, and then statistics on the single merged and mapped BAM file), so we should play around with the column configuration to help we visualise the best way to initially evaluate our data.
The bulk of the MultiQC report is made up of per-tool summary plots (e.g., barcharts, linecharts etc.). Most of these will be interactive, we can hover over lines and bars to get more specific numbers of each plot. However the visualisations are aimed at helping we identify possible outliers that may represent failed samples or libraries.
Evaluating how good the data is and how well the run went will vary depending on the dataset and the options selected. However the nf-core/eager tutorials (https://nf-co.re/eager/usage#tutorial—how-to-set-up-nf-coreeager-for-pathogen-genomics) have a good overview of questions we can ask from our MultiQC report to see whether our data is good or not. We shamelessly copy these questions here (as the overlap authors of both the the nf-core/eager documentation and this text book is rather high).
Once completed, we can try going through the MultiQC report the command we executed above, and compare against the questions below. Keep in mind we have a sample N of two, so many of the questions in regards to identifying ‘outliers’ may be difficult.
The following questions are by no means comprehensive, but rather represent the ‘typical’ questions the nf-core/eager developers asked of their own data and reports. However they can act as a good framework for thinking critically about our own results.
General Stats Table
- Do we see the expected number of raw sequencing reads (summed across each set of FASTQ files per library) that was requested for sequencing?
- Does the percentage of trimmed reads look normal for aDNA, and do lengths after trimming look short as expected of aDNA?
- Does the Endogenous DNA (%) columns look reasonable (high enough to indicate we have received enough coverage for downstream, and/or do we lose an unusually high reads after filtering )
- Does ClusterFactor or ‘% Dups’ look high (e.g. >2 or >10% respectively - high values suggesting over-amplified or badly preserved samples i.e. low complexity; note that genome-enrichment libraries may by their nature look higher)
- Do we see an increased frequency of C>Ts on the 5’ end of molecules in the mapped reads?
- Do median read lengths look relatively low (normally <= 100 bp) indicating typically fragmented aDNA?
- Does the % coverage decrease relatively gradually at each depth coverage, and does not drop extremely drastically
- Does the Median coverage and percent >3x (or whatever we set) show sufficient coverage for reliable SNP calls and that a good proportion of the genome is covered indicating we have the right reference genome?
- Do we see a high proportion of % Hets, indicating many multi-allelic sites (and possibly presence of cross-mapping from other species, that may lead to false positive or less confident SNP calls)?
FastQC (pre-AdapterRemoval)
- Do we see any very early drop off of sequence quality scores suggesting problematic sequencing run?
- Do we see outlier GC content distributions?
- Do we see high sequence duplication levels?
AdapterRemoval
- Do we see high numbers of singletons or discarded read pairs?
FastQC (post-AdapterRemoval)
- Do we see improved sequence quality scores along the length of reads?
- Do we see reduced adapter content levels?
Samtools Flagstat (pre/post Filter)
- Do we see outliers, e.g. with unusually low levels of mapped reads, (indicative of badly preserved samples) that require downstream closer assessment?
DeDup/Picard MarkDuplicates
- Do we see large numbers of duplicates being removed, possibly indicating over-amplified or badly preserved samples?
MALT (metagenomics only)
- Do we have a reasonable level of mappability?
- Somewhere between 10-30% can be pretty normal for aDNA, whereas e.g. <1% requires careful manual assessment
- Do we have a reasonable taxonomic assignment success?
- we hope to have a large number of the mapped reads (from the mappability plot) that also have taxonomic assignment
PreSeq (genomics only)
- Do we see a large drop off of a sample’s curve away from the theoretical complexity? If so, this may indicate it’s not worth performing deeper sequencing as we will get few unique reads (vs. duplicates that are not any more informative than the reads you’ve already sequenced)
DamageProfiler (genomics only)
- Do we see evidence of damage on the microbial DNA (i.e. a % C>T of more than ~5% in the first few nucleotide positions?) ? If not, possibly our mapped reads are deriving from modern contamination
QualiMap (genomics only)
- Do we see a peak of coverage (X) at a good level, e.g. >= 3x, indicating sufficient coverage for reliable SNP calls?
MultiVCFAnalyzer (genomics only)
- Do we have a good number of called SNPs that suggest the samples have genomes with sufficient nucleotide diversity to inform phylogenetic analysis?
- Do we have a large number of discarded SNP calls?
- Are the % Hets very high indicating possible cross-mapping from off-target organisms that may confounding variant calling?
As above evaluating these outputs will vary depending on the data and or pipeline settings, and very much. However the extensive output documentation (https://nf-co.re/eager/2.4.7/output) of nf-core/eager can guide we through every single table and plot to assist we in continuing any type of ancient DNA project, assisted by fun little cartoony schematic diagrams (Figure 17.3)!
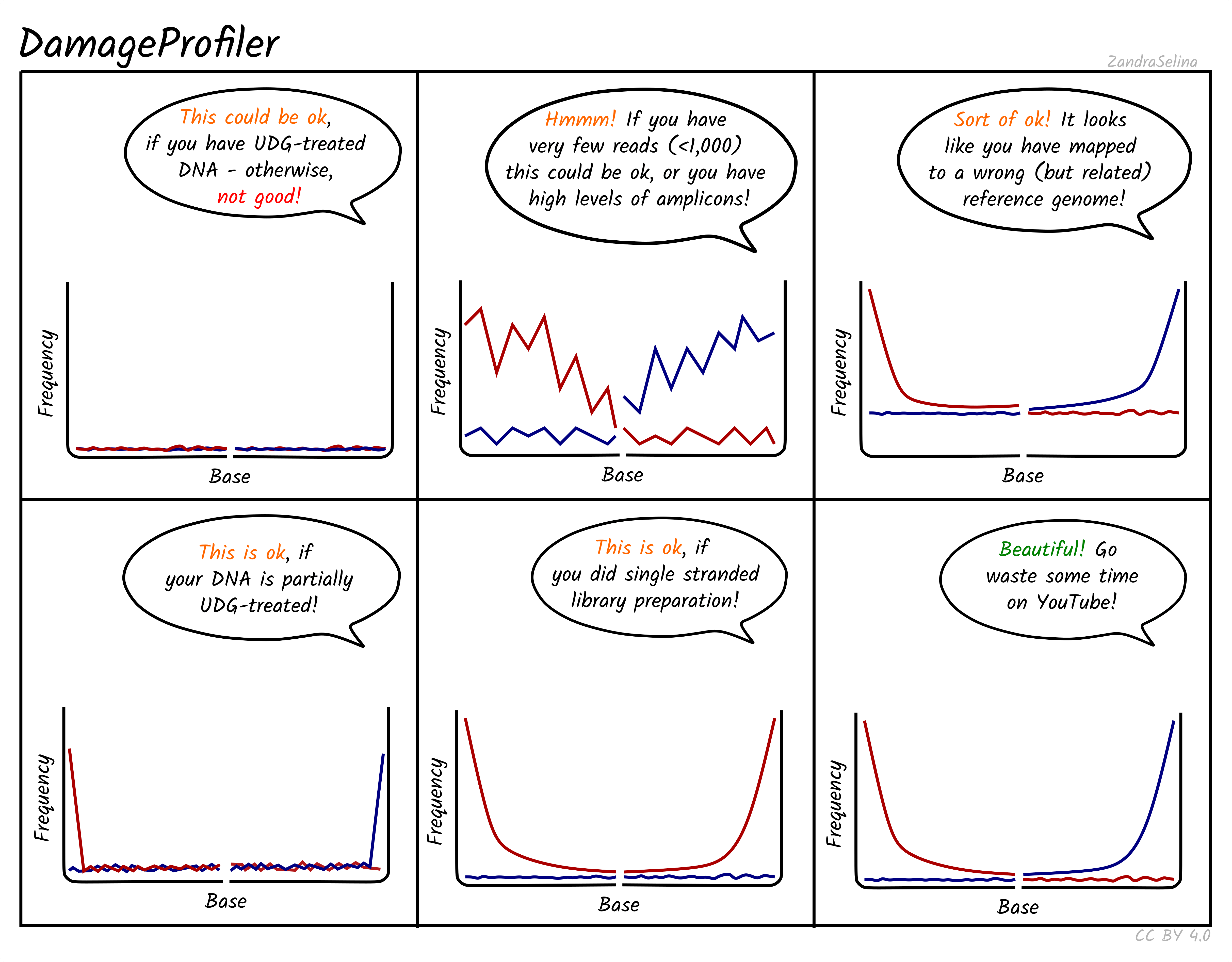
DamageProfiler (Neukamm et al. 2021) in different contexts. The six panels show different types of ancient DNA damage line-plots from having no damage (flat red/blue lines), however with a speech bubble noting that if the library was UDG treated, that the flat lines might be valid, all the way to the ‘classic’ ancient DNA damage plot with both red and blue lines showing an exponential decay from the ends of reads to the middle, with a motivational speech bubble. Source: Zandra Fagernäs, CC-BY 4.0 via nf-core/eager documentation (https://nf-co.re/eager/output).
17.3.5 Clean up
Before continuing onto the next section of this chapter, we will need to deactivate from the conda environment.
conda deactivate If the nf-core/eager run is still not finished, we should enter the screen session with screen -r eager, press ctrl + c until we drop to the prompt, and type exit.
Then may also need to delete the contents of the eager directory if we are low on hard-drive space.
cd /<path>/<to>/ancient-metagenomic-pipelines/eager
rm -r * .nex*17.4 What is aMeta?
For this chapter’s exercises, if not already performed, we will need to create the special aMeta conda environment and activate the environment.
While nf-core/eager is a solid pipeline for microbial genomics, and can also perform metagenomic screening via the integrated HOPS pipeline (Hübler et al. 2019) or Kraken2 (Wood et al. 2019), in some cases we may wish to have a more accurate and resource efficient pipeline In this section, we will demonstrate an example of using aMeta, a Snakemake workflow proposed by Pochon et al. (2022) that aims to minimise resource usage by combining both low-resource requiring k-mer based taxonomic profiling as well as accurate read-alignment (Figure 17.4).
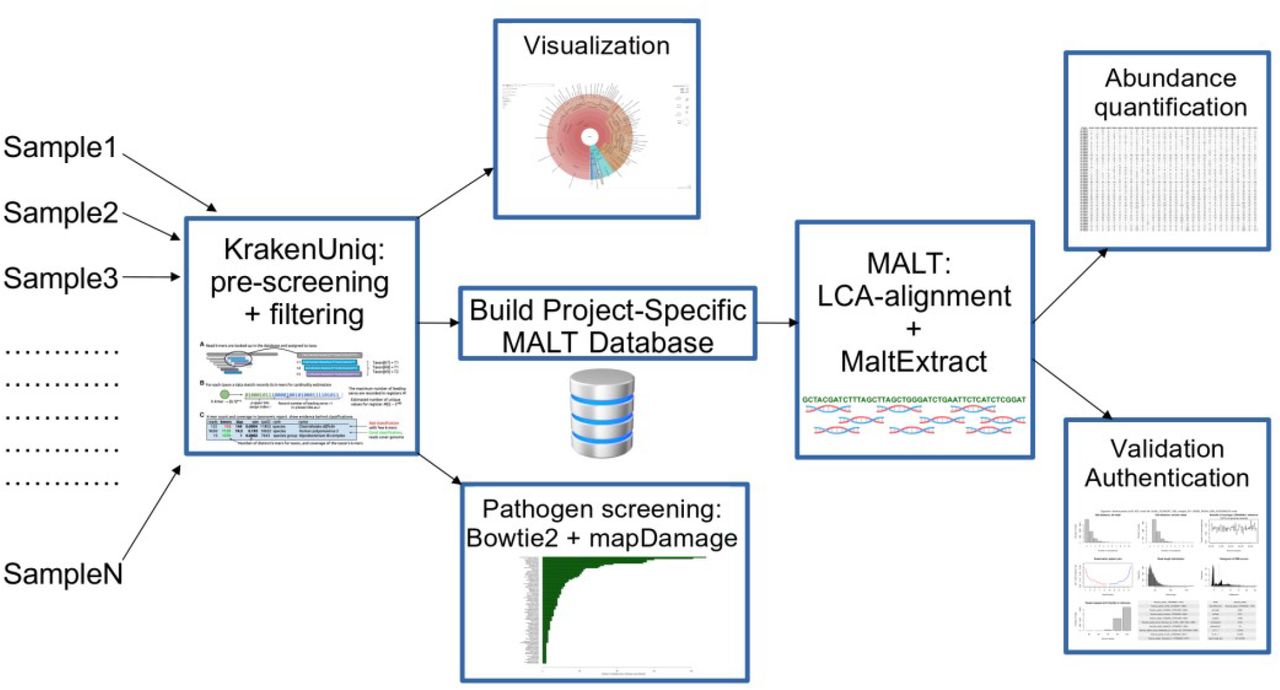
KrakenUniq. For every hit that the reads match inside this database, then sees the genome of that hit then constructed into a MALT database where the reads undergo a mapping step to generate alignments, a lowest-common-ancestor (LCA) algorithm step to refine taxonomic assignments, and ancient DNA authentication statistics generation
Rather than the very computationally heavy HOPS pipeline (Hübler et al. 2019), that requires extremely large computational nodes with large RAM (>1 TB) to load MALT databases into memory, aMeta does this via a two step approach. Firstly it uses KrakenUniq (a k-mer based and thus memory efficient method, Breitwieser et al. 2018) to do a screening of sequencing reads against a broad generalised microbial database. Once all the possible taxa have been detected, aMeta will then make a new database of just the genomes of the taxa that were reported from KrakenUniq (i.e. a project-specific database) but using MALT. Then aMeta will useMALTon thus much reduced database to perform computationally much heavier alignment against the reference genomes and LCA taxonomic reassignment. The output fromMALTis then sent to theMaltExtract` program of the HOPS pipeline for ancient DNA authentication statistics.
17.4.1 Running aMeta
In this tutorial we will try running the small test data that comes with aMeta.
aMeta has been written in Snakemake, which means running the pipeline has to be installed in a slightly different manner to the nextflow pull command that can be used for nf-core/eager.
Make sure you have followed the instructions in the Before You Start Chapter for cloning the aMeta GitHub repository to the ancient-metagenomic-pipelines/ directory. Once we have done this, we can make sure we are in the aMeta directory, if not already.
cd /<path>/<to>/ancient-metagenomic-pipelines/aMetaAnd activate the dedicated aMeta conda environment.
conda activate aMetaAs aMeta also includes tools that normally require very large computational resources that cannot fit on a standard laptop, we will instead try to re-use the internal very small ‘fake’ data the aMeta developers use to test the pipeline.
The following steps are already performed up for Students in the summer schools as, particularly the set up conda envs will take a very long time!
If you are doing this chapter self-guided, it is critical to perform the set up steps in the collapsed ‘Self guided’ block below!
We don’t have to worry about trying to understand exactly what the following commands are doing, they will not be important for the rest of the chapter. However generally the commands try to pull all the relevant software (via conda), make a fake database and download other required files, and then reconstruct the basic directory and file structure required for running the pipeline.
## Change into ~/.test to set up all the required test resources (Databases etc.)
cd .test/
## Set up conda envs
## If we can an error about a 'non-default solver backend' the run `conda config --set solver classic` and re-start the command
## If we have installed the standalone mamba, change --conda-frontend to mamba
snakemake --use-conda --show-failed-logs -j 2 --conda-cleanup-pkgs cache --conda-create-envs-only -s ../workflow/Snakefile --conda-frontend conda
source $(dirname $(dirname $CONDA_EXE))/etc/profile.d/conda.sh
## If we had to change the solver back to classic, we can switch to libmamba with `conda config --set solver libmamba`
## Build dummy KrakenUniq database
env=$(grep krakenuniq .snakemake/conda/*yaml | awk '{print $1}' | sed -e "s/.yaml://g")
conda activate $env
krakenuniq-build --db resources/KrakenUniq_DB --kmer-len 21 --minimizer-len 11 --jellyfish-bin $(pwd)/$env/bin/jellyfish
conda deactivate
## Get Krona taxonomy tax dump
env=$(grep krona .snakemake/conda/*yaml | awk '{print $1}' | sed -e "s/.yaml://g" | head -1)
conda activate $env
cd $env/opt/krona
./updateTaxonomy.sh taxonomy
cd -
conda deactivate
## Adjust malt max memory usage
env=$(grep hops .snakemake/conda/*yaml | awk '{print $1}' | sed -e "s/.yaml://g" | head -1)
conda activate $env
version=$(conda list malt --json | grep version | sed -e "s/\"//g" | awk '{print $2}')
cd $env/opt/malt-$version
sed -i -e "s/-Xmx64G/-Xmx3G/" malt-build.vmoptions
sed -i -e "s/-Xmx64G/-Xmx3G/" malt-run.vmoptions
cd -
conda deactivate
touch .initdb
## Run a quick test and generate the report (you can open this to check it looks like everythin was generated)
snakemake --use-conda --show-failed-logs --conda-cleanup-pkgs cache -s ../workflow/Snakefile $@ --conda-frontend conda -j 4
snakemake -s ../workflow/Snakefile --report --report-stylesheet ../workflow/report/custom.css --conda-frontend conda
## Now we move back into the main repository where we can symlink all the database files back to try running our 'own' test
cd ../
cd resources/
ln -s ../.test/resources/* .
cd ../config
mv config.yaml config.yaml.bkp
mv samples.tsv samplest.tsv.bkp
cd ../
ln -s .test/data/ .
ln -s .test/.snakemake/ . ## so we can re-use conda environments from the `.test` directory for the summer school run
## Again get the taxonomy tax dump for Krona, but this time for a real run
## Make sure you're now in the root directory of the repository!
env=$(grep krona .test/.snakemake/conda/*yaml | awk '{print $1}' | sed -e "s/.yaml://g" | head -1)
conda activate $env
cd $env/opt/krona
./updateTaxonomy.sh taxonomy
cd -
conda deactivate
## And back to the root of the repo for practising aMeta properly!
cd ../Now hopefully we can forget all this, and imagine we are running data though aMeta as you would normally from scratch.
OK now aMeta is all set up, we can now simulate running a ‘real’ pipeline job!
17.4.2 aMeta configuration
First, we will need to configure the workflow. First, we need to create a tab-delimited samples.tsv file inside aMeta/config/ and provide the names of the input fastq-files. In a text editor (e.g. nano), write the following names paths in TSV format.
sample fastq
bar data/bar.fq.gz
foo data/foo.fq.gzMake sure when copy pasting into our test editor, tabs are not replaced with spaces, otherwise the file might not be read!
aMeta (v1.0.0) currently only supports single-end or pre-merged- data only!
Then we need to write a config file. This tells aMeta where to find things such as database files and other settings.
A minimal example config.yaml files can look like this. This includes specifying the location the main samplesheet, which points to a TSV file that contains all the FASTQs if the samples we want to analyse, and paths to all the required database files and reference genomes you may need. These paths and settings go inside the config.yaml file that must be placed inside inside aMeta/config/.
Make a configuration file with your text editor of choice (e.g. nano).
samplesheet: "config/samples.tsv"
krakenuniq_db: resources/KrakenUniq_DB
bowtie2_db: resources/ref.fa
bowtie2_seqid2taxid_db: resources/seqid2taxid.pathogen.map
pathogenomesFound: resources/pathogenomesFound.tab
malt_nt_fasta: resources/ref.fa
malt_seqid2taxid_db: resources/KrakenUniq_DB/seqid2taxid.map
malt_accession2taxid: resources/accession2taxid.map
ncbi_db: resources/ncbi
n_unique_kmers: 1000
n_tax_reads: 200Once this config file is generated, we can start the run.
As this is only a dummy run (due to the large-ish computational resources required for MALT), we re-use some of the resource files here. While this will produce nonsense output, it is used here to demonstrate how we would execute the pipeline.
17.4.3 Prepare and run aMeta
Make sure we’re still in the aMeta conda environment, and that we are still in the main aMeta directory with the following.
conda activate aMeta
cd /<path/<to>/ancient-metagenomic-pipelines/aMeta/Finally, we are ready to run aMeta, where it will automatically pick up our config and samplesheet file we placed in config/!
## change conda-frontend to mamba if we set this up earlier!
snakemake --snakefile workflow/Snakefile --use-conda -j 10 --conda-frontend condaWe can modify -j to represent the number of available CPUs we have on our machine.
If we are using conda is the frontend this could be quite slow!
Firstly we’ll have displayed on the console a bunch of messages about JSON schemas, building DAG of jobs and downloading installing of conda environments. We will know the pipeline has started when we get green messages saying things like Checkpoint and Finished job XX.
We’ll know the job is completed once we get the following messages
Finished job 0.
31 of 31 steps (100%) done
Complete log: .snakemake/log/2023-10-05T155051.524987.snakemake.log17.4.4 aMeta output
All output files of the workflow are located in aMeta/results directory. To get a quick overview of ancient microbes present in our samples we should check a heatmap in results/overview_heatmap_scores.pdf.
If running during the summer school, you can use the following command to open the PDF file from the command line.
evince results/overview_heatmap_scores.pdf
The heatmap demonstrates microbial species (in rows) authenticated for each sample (in columns) (Figure 17.5).
The colors and the numbers in the heatmap represent authentications scores, i.e. numeric quantification of eight quality metrics that provide information about microbial presence and ancient status.
The authentication scores can vary from 0 to 10, the higher is the score the more likely that a microbe is present in a sample and is ancient.
Typically, scores from 8 to 10 (red color in the heatmap) provide good confidence of ancient microbial presence in a sample. Scores from 5 to 7 (yellow and orange colors in the heatmap) can imply that either: (a) a microbe is present but not ancient, i.e. modern contaminant, or (b) a microbe is ancient (the reads are damaged) but was perhaps aligned to a wrong reference, i.e. it is not the microbe we think about.
The former is a more common case scenario.
The latter often happens when an ancient microbe is correctly detected on a genus level but we are not confident about the exact species, and might be aligning the damaged reads to a non-optimal reference which leads to a lot of mismatches or poor evennes of coverage. Scores from 0 to 4 (blue color in the heatmap) typically mean that we have very little statistical evidence (very few reads) to claim presence of a microbe in a sample.
To visually examine the seven quality metrics
- Deamination profile
- Evenness of coverage
- Edit distance (amount of mismatches) for all reads
- Edit distance (amount of mismatches) for damaged reads
- Read length distribution
- PMD scores distribution
- Number of assigned reads (depth of coverage)
- average nucleotide identity (ANI)
Corresponding to the numbers and colors of the heatmap, one can find them in results/AUTHENTICATION/sampleID/taxID/authentic_<Sample>_<sampleID>.trimmed.rma6_<TaxID>_<taxID>.pdf for each sample sampleID and each authenticated microbe taxID. An example of such quality metrics is shown below in Figure 17.6.
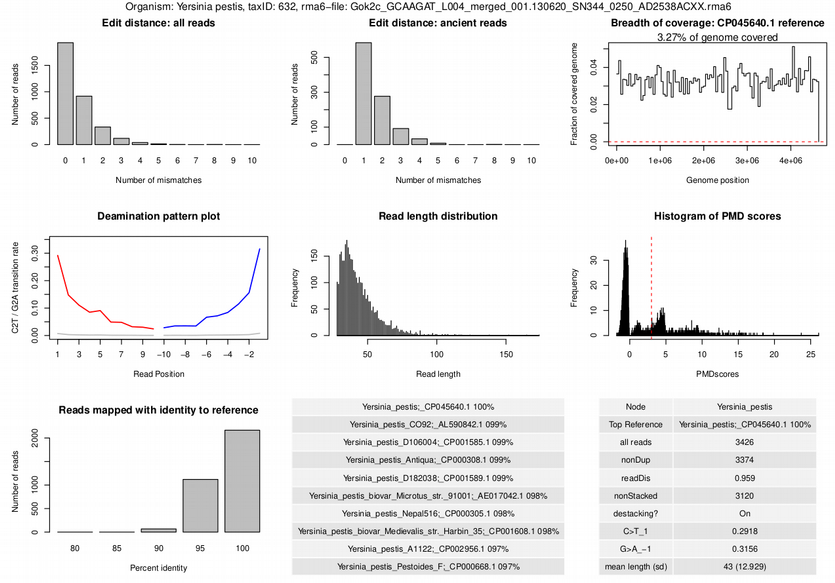
In our test data, what score does the sample ‘foo’ for the hit against Yersinia pestis? Is this a good score?
Inspect the results AUTHENTICATION/xxx/authentic_Sample_foo_*.pdf file What could have contributed to this particular score?
Hint: Check Supplementary File 2, section S5 of (Pochon et al. 2022) for some hints.
The sample foo gets a score of 4. This is a low score, and indicates that aMeta is not very confident that this is a true hit. The metrics that contribute to this score are:
- Edit distance all reads (+1)
- Deamination plot (+2)
- Reads mapped with identity (+1),
17.4.5 Clean up
Before continuing onto the next section of this chapter, we will need to remove the output files, and deactivate from the conda environment.
rm -r results/ log/
## You can also optionall remove the conda environments if we are running out of space
# rm -r .snakemake/ .test/.snakemake
conda deactivate 17.5 What is nf-core/mag?
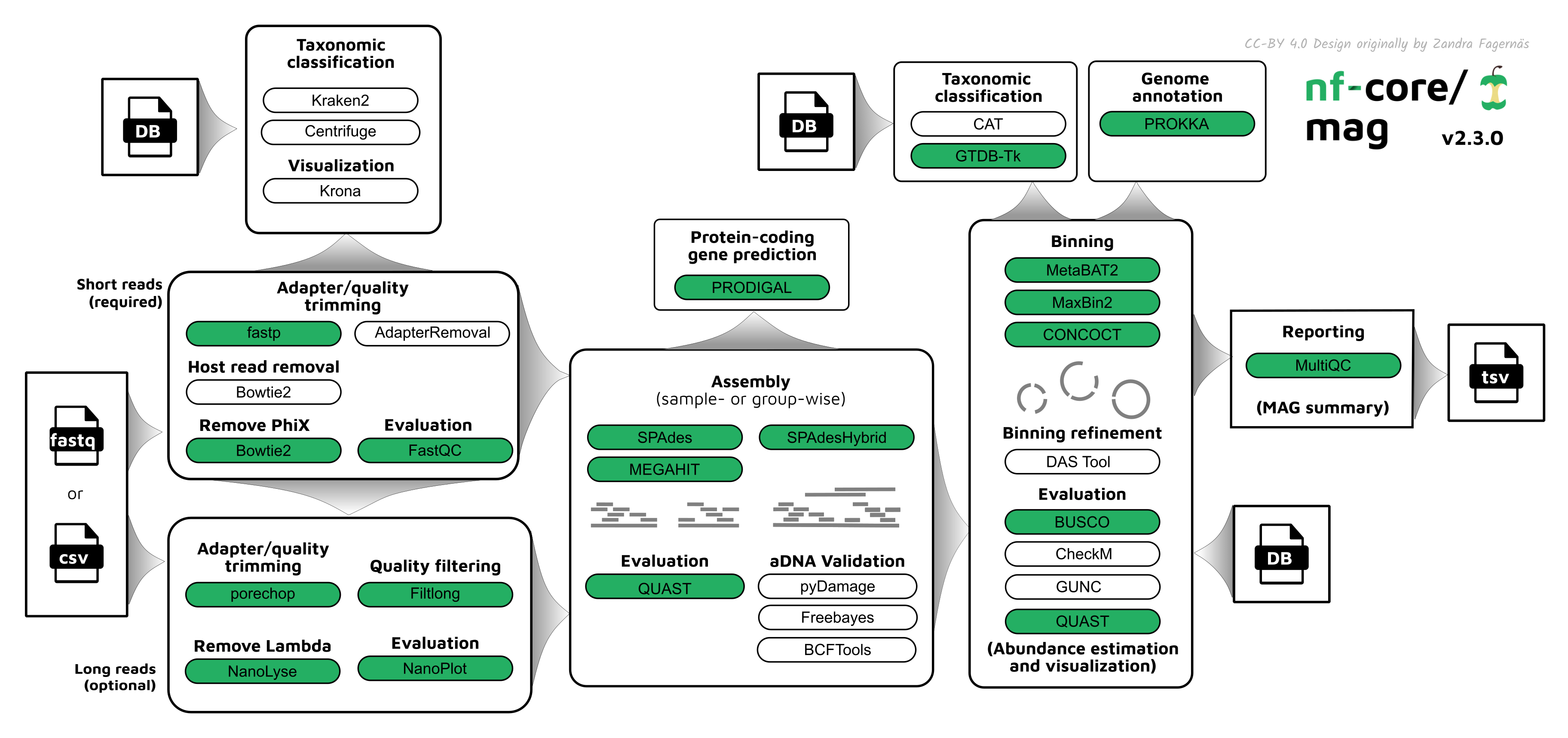
nf-core/mag (Krakau et al. 2022) is another Nextflow best-practise pipeline for the de novo assembly, binning, and annotation of metagenomes (Figure 17.7). While it was originally designed for modern metagenomes (with a focus on optional support co-assembly with Nanopore long reads), it has since been updated to include specific functionality and parameters for ancient metagenomes too.
MultiQC.
nf-core/mag covers the same major steps as we will have run if we followed the chapter on De Novo assembly.
Firstly, as with nf-core/eager, nf-core/mag do some initial sequencing QC and cleanup with fastp (Chen et al. 2018) or AdapterRemoval (Schubert et al. 2016), running FastQC (Andrews 2010) before and after this step to make sure that adapters and overly short reads have been removed. It can then optionally do basic taxonomic classification of raw reads with Kraken2 (Wood et al. 2019) or Centrifuge (Kim et al. 2016). The processed reads then ungo assembly with MEGAHIT (Li et al. 2015), metaSPAdes (Nurk et al. 2017), or SPAdeshybrid (Antipov et al. 2016) (the latter being for combining short and long reads), with contig annotation with Prodigal (Hyatt et al. 2010) to get gene prediction and assembly statistics with QUAST. A unique feature of nf-core/mag over other de novo assembly pipelines is an aDNA validation and correction step: contigs under go ‘reference free’ damage profiling using pyDamage (Borry et al. 2021), and then remaining damage bases that are mistakenly incorporated by assemblies are corrected using freebayes (Garrison and Marth 2012) and BCFTools. Contigs then optionally grouped into genomic ‘bins’ with a range of metagenomic binners (MetaBAT2 (Kang et al. 2019), MaxBin2 (Wu et al. 2015), and CONCOCT (Alneberg et al. 2014)), as well as also optional binning refinement with DAS Tool (Sieber et al. 2018). Bins are then re-annotated with Prokka (Seemann 2014), taxonomically classified with either CAT (Meijenfeldt et al. 2019) or GTDB-Tk (Chaumeil et al. 2022) Evaluation of the resulting bins is carried out with BUSCO (Simão et al. 2015), CheckM (Parks et al. 2015), GUNC (Orakov et al. 2021) and/or QUAST (Gurevich et al. 2013) to assess the bin completeness. And, as with all nf-core pipelines, all the results are wrapped up into a MultiQC report. Detailed documentation can be see on the nf-core/mag parameters (https://nf-co.re/mag/2.3.2/parameters) and output (https://nf-co.re/mag/2.3.2/docs/output pages.
Why do we need ancient DNA specific steps in a de novo assembly pipeline?
Firstly we need a method of validating that contigs are from an ancient genome (in nf-core/mag, damage patterns are generated by pyDamage).
Secondly, to correct for falsely included damage bases in contigs in some contexts. As previously stated a previous chapter by Alex Hübner:
In our group we realised that the gene sequences that were annotated on the contigs of MEGAHIT tended to have a higher number of nonsense mutations compared to the known variants of the genes. After manual inspection, we observed that many of these mutations appeared because MEGAHIT chose the damage-derived T allele over the C allele or the damage-derived A allele over a G allele (see Klapper et al. 2023, fig. S1).
To overcome this issue, my colleagues Maxime Borry, James Fellows Yates and I developed a strategy to replace these damage-derived alleles in the contig sequences. This approach consists of aligning the short-read data against the contigs, performing genotyping along them, and finally replacing all alleles for which we have strong support for an allele that is different from the one selected by MEGAHIT.
See the De novo assembly chapter for full context.
17.5.1 Running nf-core/mag
First re-activate the chapter’s conda environment.
conda activate ancient-metagenomic-pipelinesIn this tutorial, we will use the same data and aim to replicate the steps taken in the De Novo assembly chapter. It is important to note that generally, de novo assembly, is very computational intensive. In many cases it will not be able to run a de novo assembly on a standard laptop, therefore some of the parameters used here have been tweaked to get the pipeline runable on powerful laptop level machines.
This tutorial will re-use the data from the de novo assembly chapter. If we have not-run that practical, or deleted the data, pleas follow the instructions at the top of the de novo assembly page to download the denovo-assembly.tar.gz file and unpack it into the denovo-assembly directory.
Make sure once we’ve finished, change back into this chapter’s directory with the following.
cd /<path>/<to>/ancient-metagenomic-pipelinesThe manual steps of the previous chapter included:
- Read clean up with
fastp, - Assembly with
MEGAHIT, - Ancient DNA assessment and correction with
freebayesandpyDamage - Binning using
MetaBAT2andMaxBin2(originally using theMetaWRAPpipeline (Uritskiy et al. 2018)), - Bin assessment with
CheckM - Contig taxonomic classification with
MMSeqs2(Steinegger and Söding 2017) via theGTDBdatabase - Genome annotation with
Prokka
So, what command would we use to recreate these?
## 🛑 Don't run this yet! 🛑
nextflow run nf-core/mag -r 2.3.2 \
-profile test,docker \
--input '../../denovo-assembly/seqdata_files/*{R1,R2}.fastq.gz' --outdir ./results \
--ancient_dna --binning_map_mode own \
--binqc_tool checkm --checkm_db data/checkm_data_2015_01_16/ \
--centrifuge_db false --kraken2_db false --skip_krona --skip_spades --skip_spadeshybrid --skip_maxbin2 --skip_concoct --skip_prodigal --gtdb false --busco_reference false In fact, nf-core/mag will do most of the steps for us! In this case, we mostly just need to deactivate most of the additional steps (the last line). Otherwise, as with eager we have done the following:
- Tell
Nextflowto run the nf-core/mag pipeline with version 2.3.2 - Specify which computing and software environment to use with
-profile- In this case we are running locally so we don’t specify a computing environment (such as a configuration for an institutional HPC)
- We use
dockeras our container engine, which downloads all the software and specific versions needed for nf-core/mag in immutable ‘containers’, to ensure nothing get broken and is as nf-core/mag expects
- Provide the various paths to the input files - the raw FASTQ files and the output directory
- Specify we want to run the aDNA mode with mapping of reads back to the original contigs rather than for co-assembly
The aDNA mode of nf-core/mag only supports mapping reads back to the original contigs. The other mapping modes are when doing co-assembly, where we assume there are similar organisms across all our metagenomes, and wish to map reads from all samples in a group to a single sample’s contig (for example). Doing this on aDNA reads would risk making false positive damage patterns, as the damaged reads may derive from reads from a different sample with damage, that are otherwise not present in the sample used for generating the given contig.
5 . Specify we want to generate completeness statistics with CheckM (rather than BUSCO), with the associated pre-downloaded database
Most nf-core pipelines will actually download reference databases, build reference indices for alignment, and in some cases reference genomes for we if we do not specify them as pipeline input. These download and reference building steps are often very time consuming, so it’s recommended that once we’ve let the database download it once, we should move the files somewhere safe and we can re-use in subsequent pipeline runs.
6 . Skip a few steps that are either ‘nice to have’ (e.g. read taxonomic classification), or require large computational resources (e.g.metaSPAdes, CONCOCT or BUSCO)
The steps we are skipping are: host/arefact removal (Bowtie2 removal of phiX), taxonomic classification of reads (centrifuge, kraken2, krona), the additional assemblers (metaSPAdes and SPAdeshybrid, as these require very large computational resources), additional binners (MaxBin2 and CONCOCT, while they are used in the de novo assembly chapter, they take a long time to run), ship raw contig annotation (with Prodigal, as we do this at the bin level), and contig taxonomic classification (with GTDB and BUSCO as they require very large databases).
With this we are almost ready for running the pipeline!
17.5.2 Configuring Nextflow pipelines
Before we execute the command however, we need to again consider about the resource requirements. As described above, particularly de novo assembly (but also many other bioinformatic analyses) require a lot of computational resources, depending on the type of data and analyses we wish to run.
For most pipelines we must tweak the resource requirements to ensure a) they will fit on our cluster, and b) will run optimally so we don’t under- or over- use our computational resources, where latter will either make ourselves (takes too long), or our cluser/server system administrators (blocks other users) unhappy!
While nf-core pipelines all have ‘reasonable default’ settings for memory, CPU, and time limits, they will not work in all contexts. Here we will give a brief example of how to tweak the parameters of the pipeline steps so that they can run on our compute node or laptop.
For Nextflow, we must make a special ‘config’ that defines the names of each step of the pipeline we wish to tweak and the corresponding resources.
For this our nf-core/mag run we will need to open our text editor an empty file called custom.config, and copy and paste the contents of the next code block.
process {
withName: FASTP {
cpus = 8
}
withName: BOWTIE2_PHIX_REMOVAL_ALIGN {
cpus = 8
}
withName: BOWTIE2_ASSEMBLY_ALIGN {
cpus = 8
}
withName: PYDAMAGE_ANALYZE {
cpus = 8
}
withName: PROKKA {
cpus = 4
}
withName: MEGAHIT {
cpus = 8
}
withName: CHECKM_LINEAGEWF{
memory = 24.GB
cpus = 8
ext.args = "--reduced_tree"
}
}Here we set the number of CPUs for most tools to a maximum of 8, and then we limit the amount of memory for CheckM to 24 GB (down from the default (https://github.com/nf-core/mag/blob/66cf53aff834d2a254b78b94fc54cd656b8b7b57/conf/base.config#L37-L41) of 36 GB - which exceeds most laptop memory). We also give a specifc CheckM flag command to use a smaller database.
We can save this custom.config file, and supply this to the Nextflow command with the Nextflow parameter (-c). The good thing about the config file, is we can re-use across runs on the same machine! So set once, and never think about it again.
And with that, we can run our nf-core/mag pipeline!
As recommended in the nf-core/eager section above, start a screen session.
screen -R magThen execute the command.
nextflow run nf-core/mag -r 2.3.2 \
-profile test,docker \
--input '../../denovo-assembly/seqdata_files/*{R1,R2}.fastq.gz' --outdir ./results \
--ancient_dna --binning_map_mode own \
--binqc_tool checkm --checkm_db data/checkm_data_2015_01_16/ \
--centrifuge_db false --kraken2_db false --skip_krona --skip_spades --skip_spadeshybrid --skip_maxbin2 --skip_concoct --skip_prodigal --gtdb false --busco_reference false \
-c custom.configAgain as with nf-core/eager when the run starts, we see a long list of process lines with progress bars slowly appearing over time. If in a screen session, we can press ctrl + a and then [ to access a ‘navigation’ (called ‘copy’) mode, then use our arrow keys on our keyboard to scroll up and down. To quit this mode just press q.
The pipeline run above should take around 20 minutes or so to complete.
Configuration files don’t need be personal nor custom!
We can also generate a HPC / server specific profile which can be used on all nf-core pipelines (and even our own!).
See nf-co.re/configs (https://nf-co.re/configs) for all existing institutional configs
Why should you make sure to configure pipeline that use a workflow manager in the backend?
The primary reason for optimising the configuration of workflow managers is efficiency.
By correctly configuring your can ensure:
- A maximal number of jobs can run in parallel
- Runs are not killed (and repeatedly retried) if they request too many resources
- Can reuse files across runs, such as databases and software containers
amongst others.
17.5.3 nf-core/mag output: results files
As with nf-core/eager, we will have a results directory containing many folders representing different stages of the pipeline, and a MultiQC report.
Relevant directories for evaluting our assemblies are as follows:
QC_shortreads/- Contains various folders from the sequencing QC and raw read processing (e.g.,
FastQC,fastp/AdapterRemoval, host-removal results etc.)
- Contains various folders from the sequencing QC and raw read processing (e.g.,
Taxonomy/- If we have not already performed taxonomic classification of our reads, the output of the
Kraken2orCentrifugeintegrated steps of nf-core/mag will be deposited here, including an optional Krona piechart - If activated, the contig-level taxonomic assignments will be deposited here (e.g., from
CATorGTDB)
- If we have not already performed taxonomic classification of our reads, the output of the
Assembly/- This directory will contain one directory per chosen assembler which will contain per-sample output files for each assembly.
- Within each of the assembler sub directories, a QC folder will contain the
QUASToutput
Ancient_DNA- This folder will contain the output of
pyDamagecontig authentication to allow we to select or filter for just putatively ancient contigs
- This folder will contain the output of
variant_calling- This directory will contain the ‘damage-corrected’ contigs that will be used in downstream steps of the pipeline, if the
aDNAmode is turned on
- This directory will contain the ‘damage-corrected’ contigs that will be used in downstream steps of the pipeline, if the
GenomeBinning/- This directory will contain all the output from each of any of the selected contig binning tools. As well as the binned FASTAs, we also have read-depth statistics from when input reads are re-mapped back to the contigs
- The directory will also contain the output from the
DASToolbinning refinement if turned on - In the
QCsub-directory of this folder, we will find the QUAST results on the genome bins (rather than raw contigs as above), as well asBUSCO/CheckM/GUNCMAG completeness estimates
Prodigal/Prokka- These directories will contain the genome annotation output of the raw contigs (
Prodigal) or from bins (Prokka)
- These directories will contain the genome annotation output of the raw contigs (
17.5.4 nf-core/mag output: run report
For MultiQC, many of the questions asked in the preprocessing section of the nf-core/eager results interpretation will also apply here.
Other than the QUAST results in the Assembly/*/QC/ or GenomeBinning/*/QC/ directories, the main table used for evaluating the output files is the bin_summary.tsv table in the GenomeBinning/ directory.
In this file we woud typically want to assess the following columns:
% Complete *columns - with the higher the number of expected domain-specific genes, normally representing a better quality of the resulting bin.# contigs (>=* bp)columns - which represnts the number of contigs at different lengths, the fewer the shorter reads and the greater the longer reads we have again suggests a better assembly.This can also be evaluated by the N50 or L75 columns (as described in the De novo assembly chapter).
fastaniandclosest_placement_taxonomy- these can tell we if our particular bin has a genome very similar to existing species in taxonomic databaseswarnings- for specific comments on each assignment
17.6 (Optional) clean-up
Let’s clean up our working directory by removing all the data and output from this chapter.
The command below will remove the /<PATH>/<TO>/ancient-metagenomic-pipelines directory as well as all of its contents.
Always be VERY careful when using rm -r. Check 3x that the path we are specifying is exactly what we want to delete and nothing more before pressing ENTER!
rm -rf /<PATH>/<TO>/ancient-metagenomic-pipelines*Once deleted we can move elsewhere (e.g., cd ~).
We can also get out of the conda environment with the following.
conda deactivateTo delete the conda environment we can use conda remove.
conda remove --name ancient-metagenomic-data --all -y17.7 Questions to think about
- Why is it important to use a pipeline for genomic analysis of ancient data?
- How can the design of pipelines such as nf-core/eager pipeline help researchers comply with the FAIR principles for management of scientific data?
- What metrics do we use to evaluate the success/failure of ancient DNA sequencing experiments? How can these measures be evaluated when using nf-core/eager for data preprocessing and analysis?